

今天小编分享的这篇paper是来自《 Genome biology》的综述,其回顾了空间转录组学中统计和机器学习方法的最新发展,总结了有用的资源。

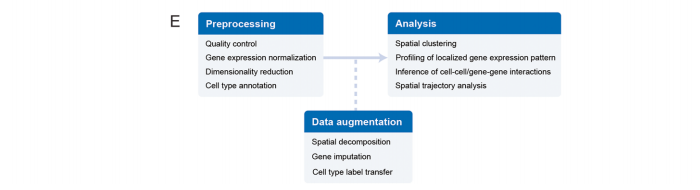
计算方法在空间转录组学研究中的应用
空间转录组学数据分析工作通常包括多个阶段:第一步是数据预处理,通常包括质量控制、基因表达标准化、降维和细胞类型注释。可以通过空间分解、基因插补和标签转移进一步提高数据的丰富性。接下来可通过空间聚类和局部基因表达模式从数据中获得生物学见解,这将进一步促进空间可变基因的识别、细胞-细胞/基因-基因相互作用的推断和空间轨迹分析。此外,空间转录组学数据可用于帮助重建scRNA-seq数据中的空间位置。
用于空间分辨转录组学数据分析的 统计和机器学习方法总结
SpatialDWLS
应用场景:Spatial decomposition(空间分解)
算法:Weighted least squares(加权最小二乘)
优点:比基准工具的精度更高、速度更快
缺点:估计稀有细胞类型比例的偏差较大
SPOTlight
应用场景:Spatial decomposition(空间分解)
算法:Seeded NMF regression(基于种子的非负矩阵因子分解回归)
优点:跨多个组织的高精度
缺点:没有将捕获的位置信息合并到模型空间分解中
RCTD
应用场景:Spatial decomposition(空间分解)
算法:Poisson distribution with MLE(泊松分布的最大似然估计)
优点:系统地模拟平台效应
缺点:假设平台效应在细胞类型之间共享
stereoscope
应用场景:Spatial decomposition(空间分解)
算法:Negative binomial distribution with MAP(具有最大后验概率的负二项分布)
优点:利用完整的表达谱而不是选定的标记基因来实现更高的准确性
缺点:需要更深的测序深度
DSTG
应用场景:Spatial decomposition(空间分解)
算法:Semi-supervised GCN(半监督图卷积神经网络)
优点:比基准工具更精确
缺点:高度依赖于建模图卷积神经网络的链接图的质量
ProximID
应用场景:Cell-cell/gene-gene interactions
S(细胞-细胞/基因-基因相互作用)
算法:Cluster label permutations(聚类标签排列)
优点:不需要物理分离 FISH 图像中的细胞
缺点:无法检测到没有物理连接的相互作用
MISTy
应用场景:Cell-cell/gene-gene interactions
S(细胞-细胞/基因-基因相互作用)
算法:Multi-view framework to dissect efects related to CCI(剖析与细胞-细胞互作相关影响的多视角框架)
优点:1.不需要细胞类型标注;2. 利用完整的表达谱
缺点:提取的相互作用不能直接视为因果关系
stLearn
应用场景:1.Cell-cell/gene-gene interactions(细胞-细胞/基因-基因相互作用);2. Spatial clustering(空间聚类);3. Cell trajectories inference(细胞轨迹推断)
算法:A toolbox containing integrated algorithms from multiple studies(包含来自多个研究的集成算法的工具箱)
优点:从原始输入到深入下游分析的简化包
缺点:仅与某些 ST 平台兼容
SVCA
应用场景:Cell-cell/gene-gene interactions
S(细胞-细胞/基因-基因相互作用)
算法:Gaussian processes(高斯过程)
优点:同时适用于RNA-seq和蛋白质组学数据
缺点:没有考虑特定于技术的噪音
GCNG
应用场景:Cell-cell/gene-gene interactions
S(细胞-细胞/基因-基因相互作用)
算法:GCN(图卷积网络)
优点:可以推断新的细胞间互作并预测新的功能基因
缺点:当应用于不同的数据集时,超参数需要重新优化
Seurat V3
应用场景:1. Gene imputation(基因插补);2. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建);3. Others(其他)
算法:Analysis pipelines with integrated algorithms(集成算法的分析管道)
优点:1. 一个全面的数据分析管道;2. 可应用于多组学数据集,包括转录组、表观基因组、蛋白质组和空间分辨率的单细胞数据
缺点:仅适用于某些类型的ST平台
LIGER
应用场景:1. Gene imputation(基因插补);2. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建)
算法:Integrative NMF(整合性非负矩阵分解)
优点:嵌入同时维护通用和特定于数据集的术语
缺点:与基准工具相比,内存密集
SpaGE
应用场景:1. Gene imputation(基因插补);2. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建)
算法:Domain adaptation model to align ST and scRNA-seq data to a common space(将 ST 和 scRNA-seq 数据对齐到公共空间的域适应模型)
优点:在大型数据集中,内存使用更少,速度比基准工具更快
缺点:模型中只包含两个数据集中的共同基因
应用场景:Gene imputation(基因插补)
算法:Autoencoder model for dimensional reduction to map ST and scRNA-seq data into a shared space(将ST和scRNA-seq数据映射到共享空间的降维自动编码器模型)
优点:1. 在细胞类型聚类方面比基准工具有更高的准确性;2. 在应用于大型数据集时,比除SpaGE以外的大多数基准工具的时间和内存使用量更少
缺点:仅适用于基于图像的测序平台的数据
gimVI
应用场景:1. Gene imputation(基因插补);2. Dimensional reduction and feature extraction(降维和特征提取)
算法:Variational autoencoders for dimensional reduction to map ST and scRNA-seq data into a shared space(具将ST和scRNA-seq数据映射到共享空间的降维变异自动编码器模型)
优点:在模型中生成平台特定的模式,以获得更好的生物可解释性
缺点:在大型数据集中比基准测试工具慢
Harmony
应用场景:1. Gene imputation(基因插补);2. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建)
算法:Maximum diversity clustering and mixture model based batch correction(基于最大多样性聚类和混合模型的批量校正)
优点:能以较高的精度估算出低丰度的基因
缺点:嵌入物缺乏生物学上的可解释性
DEEPsc
应用场景:Gene imputation(基因插补)
算法:ANN(人工神经网络)
优点:一种专门为基因插补设计的系统自适应方法
缺点:没有将空间信息合并到计算中
Trendsceek
应用场景:Identify SVGs(识别空间变异基因)
算法:Marked point process(标值点过程)
优点:不需要指定一个分布或一个感兴趣的空间区域
缺点:每次只限于单个基因,计算量大
SpatialDE
应用场景:Identify SVGs(识别空间变异基因)
算法:Gaussian process regression(高斯过程回归)
优点:可以检测时间和周期基因表达模式的SVGs识别
缺点:不识别具有不同表达模式的空间区域,计算密集型
SPARK
应用场景:1. Identify SVGs(识别空间变异基因);2. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建)
算法:Generalized linear spatial models (广义线性空间模型)
优点:1. 低误检率;2. 不需要用户对原始计数矩阵进行预处理
缺点:当应用于不同的数据集时,需要重新优化超参数(内核和权重)
SpaGCN
应用场景:1. Identify SVGs(识别空间变异基因);2. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建)
算法:GCN(图卷积网络)
优点:联合识别SVGs和空间域
缺点:没有将细胞类型信息和组织解剖结构纳入计算
SPARK-X
应用场景:1. Identify SVGs(识别空间变异基因);2. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建)
算法:Non-parametric covariance test(非参数协方差检验)
优点:与大多数基准测试工具相比,使用的时间和内存更少,低误检率,尤其是在大规模和稀疏的ST数据中
缺点:准确性因不同的相似性测量和协方差函数而异
sepal
应用场景:1. Identify SVGs(识别空间变异基因);2. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建)
算法:Difusion mode(扩散模型)
优点:可以检测不规则空间模式的基因
缺点:有CPU并行化,但没有GPU加速
GLISS
应用场景:1. Identify SVGs(识别空间变异基因);2. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建)
算法:Graph Laplacian-based model(基于图的拉普拉斯模型)
优点:不需要对空间或scRNA-seq数据进行分布式假设
缺点:需要手动或通过其他算法预先指定landmark基因
smfishhmrf-py
应用场景:
- Profle localized gene expression pattern(剖析局部基因表达模式);2. Identify SVGs(识别空间变异基因);3. Identify interactions between cell type and spatial environment(确定细胞类型和空间环境之间的相互作用)
算法:HMRF(隐马尔可夫随机场模型)
优点:可以从头识别新的空间关联亚群
缺点:仅适用于原位杂交数据集
应用场景:1. Profle localized gene expression pattern to enhance ST data resolution(提供局部基因表达模式以提高ST数据分辨率);2. Spatial clustering(空间聚类)
算法:Bayesian statistical method(贝叶斯统计方法)
优点:不需要独立的单细胞数据
缺点:仅考虑ST和Visium平台的数据中存在的邻域结构
XFuse
应用场景:Gene expression prediction from histology images(从组织学图像中预测基因表达情况)
算法:Deep generative model(深度生成模型)
优点:可用于在组织学图像中转录组水平的基因表达推断
缺点:仅适用于原位 RNA 捕获技术
Seurat V1
应用场景:1. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建);2. Gene imputation(基因插补)
算法:L1-constrained linear mode(L1约束线性模型)
优点:landmark基因的概念允许使用少量基因进行空间位置重建
缺点:需要预先计算landmark基因的位置
CSOmap
应用场景:1. Identify cell-cell/gene-gene interactions(识别细胞-细胞/基因-基因相互作用);2. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建)
算法:Reconstructs cellular spatial organization based on cell-cell afnity by ligand-receptor interactions(通过配体-受体相互作用重建基于细胞间亲和力的细胞空间组织)
优点:不需要预先定义组织形状以进行细胞间相互作用推断;不需要预先定义landmark基因集
缺点:提取的空间结构是伪空间结构
DistMap
应用场景:Construct 3D gene expression blueprint for the Drosophila embryo(构建果蝇胚胎的三维基因表达图谱)
算法:Mapping scores to measure the similarity between spatial and scRNA-seq data(映射得分来衡量空间数据和scRNA-seq数据之间的相似性)
优点:高精度,仅 84 个原位即可
缺点:基因调控可以被认为是提高模型准确性的原位方法
SpaOTsc
应用场景:1. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建);2. Cell-cell/gene-gene interactions(细胞-细胞/基因-基因相互作用);3. Identify gene pairs that potentially intercellularly regulate each other(识别可能在细胞间相互调节的基因对)
算法:Structured optimal transport model(结构化最优传输模型)
优点:1.大多数细胞只需少量基因就能精确定位;2.能够识别细胞间基因调控信息
缺点:不考虑细胞间通讯可能发生的时间延迟(包括配体的扩散时间或细胞内级联反应的时间)
novoSpaRc
应用场景:Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建)
算法:Generalized optimal-transport model(广义最优传输模型)
优点:不需要指定用于对齐的landmark基因
缺点:可以通过使用不同的损失函数来提高准确性
Tangram
应用场景:1. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建);2. Spatial decomposition(空间分解);3. Gene imputation from histology data(来自组织学数据的基因插补)
算法:Non-convex optimization by deep learning methods for spatial alignment(用深度学习方法进行空间排列的非凸优化)
优点:与基于捕获和基于图像的ST数据兼容如果无法在图像中分割细胞,则组织学基因表达预测的准确性会降低
缺点:每次只限于单个基因,计算量大
Cell2location
应用场景:1. Spatial location reconstruction for scRNA-seq data(scRNA-seq数据的空间位置重建);2. Spatial decomposition(空间分解)
算法:Hierarchical Bayesian framework(层次贝叶斯框架)
优点:能够推断每个捕获位置每种细胞类型的细胞绝对数量
缺点:用户通常不知道要预先指定的超参数
SC-MEB
应用场景:Spatially clustering(空间聚类)
算法:HMRF based on empirical Bayes(基于经验贝叶斯的隐马尔可夫随机场模型)
优点:比基准工具更快、更准确,尤其是在大型数据集中
缺点:在模型中假设一个六边形的邻域结构,可能不会对所有的ST平台保持高精确度
STAGATE
应用场景:1. Spatially clustering(空间聚类);2. Identify SVGs(识别空间变异基因)
算法:Graph attention auto-encoder(图注意力自动编码器)
优点:可以应用于三维ST数据集
缺点:两个部分的边界需要进一步细化
MULTILAYER
应用场景:1. Spatially clustering(空间聚类);2. Identify SVGs(识别空间变异基因)
算法:Agglomerative clustering of quantile normalized ST data(分位数归一化ST数据的聚类分析 )
优点:当应用于来自不同ST平台的数据时,比基准测试工具具有更高的准确性
缺点:对空间分辨率低的 ST 数据敏感
HisToGene
应用场景:Gene expression prediction from histology images(从组织学图像预测基因表达)
算法:Attention-based (vision transformer) model(基于注意力的(视觉转换器)模型)
优点:可以在捕获位置水平预测组织学图像中的基因表达
缺点:模型训练需要大量的组织样本
STARCH
应用场景:Infer copy number aberrations(推断拷贝数畸变)
算法:HMRF and HMM(隐马尔可夫随机场模型和隐马尔可夫模型)
优点:在预测空间数据集中的拷贝数变化时,比基准工具更准确
缺点:每次只限于单个基因,计算量大
Giotto
应用场景:A comprehensive toolbox for ST analysis and visualization(ST分析和可视化的综合工具箱)
算法:A toolbox containing integrated algorithms from multiple studies(一个包含多项研究的综合算法工具箱)
优点:为 ST 数据分析提供全面的管道
缺点:仅适用于部分ST平台
参考文献
Zeng Z, Li Y, Li Y, et al. Statistical and machine learning methods for spatially resolved transcriptomics data analysis[J]. Genome biology, 2022, 23(1): 1-23.
图片均来源于参考文献,如有侵权请联系删除。
首发公号:国家基因库大数据平台
Original: https://blog.csdn.net/wen05054105/article/details/125145704
Author: wen05054105
Title: 超全总结 | 用于空间分辨转录组数据分析的统计和机器学习方法
原创文章受到原创版权保护。转载请注明出处:https://www.johngo689.com/698679/
转载文章受原作者版权保护。转载请注明原作者出处!