

提示:文章写完后,目录可以自动生成,如何生成可参考右边的帮助文档
文章目录
文章地址:https://academic.oup.com/bioinformatics/article-abstract/38/3/738/6384568?redirectedFrom=fulltext
DOI:10.1093/bioinformatics/btab700
期刊:Bioinformatics(2区)
发布时间:2021年7月9日
代码:https://github.com/ddb-qiwang/scMRA-torch
前言
动机:单细胞RNA-seq (scRNA-seq)已被广泛用于解决细胞异质性。收集完scRNA-seq数据后,下一步自然是集成积累的数据,以实现细胞类型和状态的公共本体。因此,迫切需要一种有效、高效的细胞型识别方法。同时,高质量的参考数据仍然是精确标注的必要条件。但在实践中,这种有针对性的参考数据一直缺乏。为了解决这个问题,我们将多个数据集聚合到一个元数据集中,在这个元数据集上进行注释。现有的监督或半监督标注方法由于不同的排序平台而存在批处理效应,当引用数据集多时,批处理效应的严重性会增加。
结果:本文提出了一种鲁棒的基于深度学习的单细胞多参考注释器(scMRA)。在scMRA中,构建了一个知识图来表示不同数据集的细胞类型特征,并基于该知识图构建了一个图形卷积网络作为判别器。scMRA保持细胞内类型的紧密性和细胞类型在数据集中的相对位置。scMRA在将知识从多个参考数据集转移到未标记的目标领域方面具有显著的能力,因此在多参考数据的实验中,scMRA比其他最先进的标注方法具有优势。此外,scMRA可以消除批量效应。据我们所知,这是第一次尝试使用多个不充分的参考数据集来注释目标数据,相对而言,它是多个scRNA-seq数据集的最佳注释方法。
一、前言
高通量单细胞测序技术在过去的十年中发展迅速,测序数据规模从几十个提升到数千个甚至数百万个,大量的新型测序平台,如10个?基因组学铬,inDrop和Drop-seq已经出现。细胞类型的识别在单细胞rna测序数据分析中发挥着重要作用,因为精心注释的单细胞测序数据使生物学家能够进行进一步的下游分析,并提高我们对疾病的细胞机制的理解。广泛应用的细胞型注释方法首先对细胞群体进行聚类,然后通过差异表达分析找到每个聚类所特有的标记基因,最后根据其基因的本体论功能对细胞进行注释。近年来出现了大量的单细胞聚类算法。例如,Seurat使用社区发现算法Louvain来划分细胞之间的共享邻居图。SC3使用一致学习获得的细胞相似度矩阵的k-means算法来获得聚类分割。scDMFK和scziddesk利用深度自编码器同时学习细胞的降维表示和类别的生成。然而,由于标记基因在不同实验中的使用差异很大,这些方法通常泛化性能较差。此外,通过增加测序数据的规模,通过寻找标记基因来注释细胞的任务变得越来越繁重和耗时。
为了解决标记基因的问题,研究人员试图使用细胞本体数据库中的细胞类型结构来统一每个数据集的注释结果。
这种统一也直接促进了标签转移。在过去的2年中,在经过良好标注的引用数据中使用cell-type标签来标注目标数据已经成为scRNA-seq数据标注领域的一种趋势。例如,scmap将查询单元投射到参考数据上,并度量它们与参考数据中已知的聚类中心或单元之间的相似性,以执行注释。scmap模型的结构为处理大规模数据提供了一种有效的方法,但它只能利用参考数据中的cell类型信息。因此,当目标数据和参考数据来自不同的测序平台时,技术变异批处理效应可能会导致性能崩溃。
Seurat通过在标注良好的参考数据和未标注的目标数据之间寻找锚定单元对,并构建共享邻居网络,对目标数据进行监督分类和预测。然而,Seurat忽略了参考数据中特定的细胞类型知识。此外,Seurat中混合查询单元和参考单元的特征对齐和重新对齐的计算量非常大,难以克服大规模数据的复杂性。
为了克服大规模scRNA测序数据的负担,一些研究人员将目光投向了深度神经网络方法。这些方法使用非线性自动编码器嵌入序列数据到低维空间,在其中进行聚类算法。
火星和ItClust是针对单细胞测序数据进行细胞类型识别的深度聚类方法的代表。利用深度自编码器对无标记目标数据集进行预训练,并利用深度神经网络学习细胞类型标志和非线性细胞嵌入。MARS将目标数据中的单元分配给具有最近地标的单元类型。然而,仅仅通过比较目标细胞到潜在聚类中心的距离来预测细胞类型,忽略了参考数据和目标数据之间的批量效应,从而存在过拟合问题。ItClust是一种基于深度迁移学习的监督聚类和细胞型分类方法。它利用了从参考数据中获得的细胞类型特异性基因表达信息。利用细胞嵌入和聚类中心之间的距离来预测细胞类型,ItClust与MARS相似,存在批量效应和信息丢失。将参考数据和目标数据分开训练也不是最理想的,因为在训练目标数据时,从源数据中积累的知识会被遗忘。
无监督域适应(Unsupervised Domain adaptive, UDA)关注的是知识从有标记的数据集(源域)到无标记数据集(目标域)的可转移性。人们提出了各种策略来解决这个问题,包括最小化明确定义的领域差异度量和基于对抗训练的领域混淆。
UDA的目的是通过增强不同领域特征表示的领域不变性来转移知识,非常适合应用于单细胞测序数据标注任务。然而,在实际应用程序中,假设所有参考数据都来自同一个领域是不合理的。在实践中,我们总是倾向于使用尽可能多的相关且注释良好的数据集作为参考数据,以确保出现在目标数据中的所有单元格类型都可以被查询,并且目标数据的注释可以更精确。这意味着我们的参考数据更有可能从不同的平台收集,从不同的组织中测序细胞,即从多个域。将这些因素整合到域对齐中是一个更实际的问题,称为多源域适应(MSDA) ,更适合我们单细胞测序数据标注的情况。利用scRNA-seq数据的cell-type identification的特点,我们提出了一种新的标注算法scMRA。
scMRA是专门为将知识从多个良好注释的数据转换为目标未标记的数据而设计的。在训练阶段,执行了四个主要步骤,如图1所示。
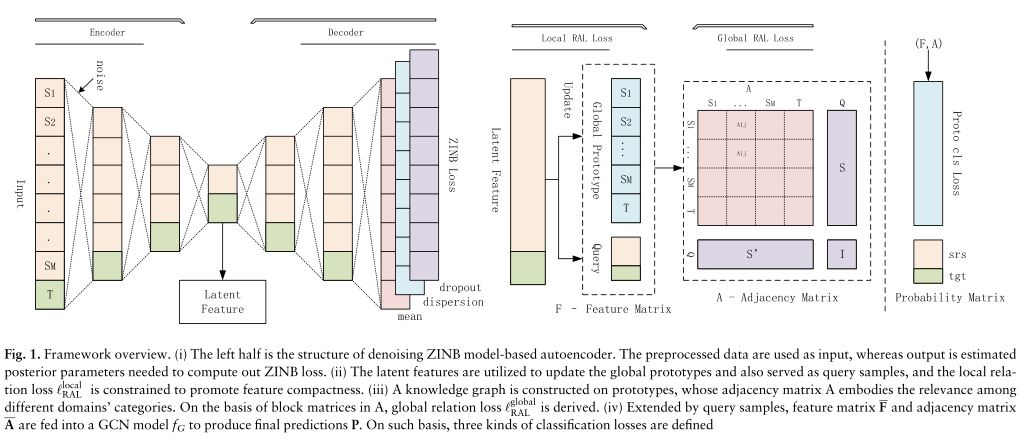

图1。框架的概述。(i)左半部分是基于ZINB模型的自动编码器的去噪结构。预处理后的数据被用作输入,而输出是估计计算ZINB损失所需的后验参数。(ii)利用潜在特征对全局原型进行更新,并作为查询样本,约束局部关系损失的局部RAL,以提高特征的紧致性。(iii)在原型上构建了一个知识图,其邻接矩阵A体现了不同领域类别之间的相关性。基于A中的块矩阵,导出了全局关系损失的全局RAL。(iv)通过查询样本扩展,将特征矩阵F和邻接矩阵A输入GCN模型fG,产生最终预测p。在此基础上,定义了三种分类损失
首先,我们通过引入一个基于ZINB模型的去噪自动编码器,将在测序阶段遭受dropout事件的测序数据进行对齐。其次,我们计算所有数据集的每个单元类型的原型表示,并使用移动平均方案更新全局原型。通过这种方式,我们减轻了这些估计的随机性,并重视跨数据集相同细胞类型的多样性。第三,在全局原型的基础上构建知识图来整合整个信息,两个原型之间的连接权值由它们的相似度决定;然后通过查询批处理中的样本扩展知识图。随后使用图形卷积网络(GCN) (Kipf和Welling, 2017)在整个扩展的图中传播特征表示,并输出每个节点的分类概率。通过GCN进行细胞识别,避免了忽略细胞间类型信息,有利于基于分类概率熵发现新的细胞类型。
最后,对损失函数进行优化,包括ZINB损失、关系对齐损失(RAL)和分类损失。RAL的优化保持了不同单元类型在数据集之间的相对位置,有助于消除批处理效应和对齐数据集。大量的仿真和真实数据实验表明,scMRA在半监督单元式标注上具有良好的无偏性能,特别是在使用多个参考数据集的情况下。
; 二、材料和方法
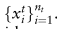
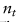
第m个参考数据
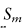
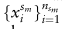
的注释良好的细胞
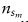
和单元格类型标签
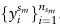
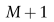
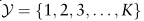
表示K种不同的细胞类型。
我们的目标是实现从参考数据到目标数据的标签转移,即通过学习共享的低维流形空间z和判别聚类函数f来标注目标细胞。
2.1基于ZINB模型的自动编码器去噪
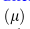
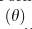
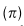
,表示0点的概率质量的权重(dropout事件的概率):
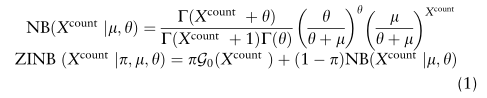
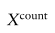
表示原始读取计数
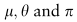
基于ZINB模型的自动编码器的损耗函数为ZINB分布的负对数似然:
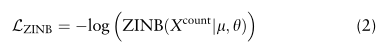
2.2知识图分类
在基于ZINB模型的去噪自动编码器的每次训练迭代中,包含所有 M+1域样本的小批量映射到低维潜在空间。
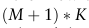

的阶数。
如果某些单元格类型在某些域中不存在,那么原型将相应地为零。
为了区分目标数据中可能出现的新细胞类型,我们根据目标细胞预测标签的熵对其进行过滤。每个批次的特征嵌入被用来更新全局原型,同时也用作查询。然后,利用全局原型和查询样例构建知识图。最后,利用GCN模型对知识图的每个节点进行信息传播和分类概率输出。这三个部分的细节如下所示。
2.2.1全球原型维护
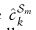
定义为所有属于cell-type标签k的细胞在Sm中的均值嵌入:
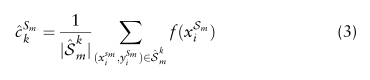
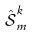
是采样Sm中所有具有cell-type标签k的细胞的集合,
f表示第2.2节介绍的基于ZINB模型去噪的自动编码器的编码器部分,它将读取计数矩阵映射到特征嵌入。
由于目标数据T无法获得地面真实信息,我们首先使用Zhang et al.(2018)提出的策略为
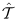
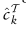
定义如下:
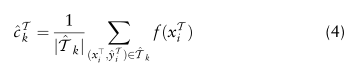
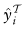
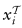
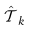
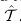
中标记为类k的样本的集合。
为了纠正小批量抽样随机性带来的估计偏差,我们用指数移动平均方案保持源域和目标域的全局原型:
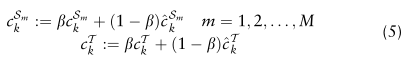
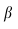
为指数衰减率,所有实验均固定为0.65。
此外,如果在一个小批处理中没有来自数据集 Sm或者 T的单个样本与标签k可用,则相应的全局原型将不会相应更新。
这种移动平均算法被广泛应用于计算机视觉领域,通过平滑全局变量来稳定训练过程。
2.2.2知识图构建
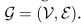
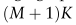
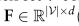
(d是特征嵌入的维度)的维数定义为全局原型的串联:
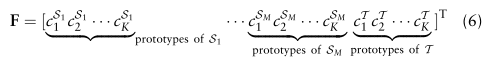
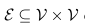
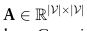
用来模拟这种关系的。
对全局原型来推导邻接矩阵
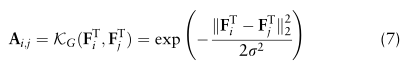
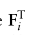
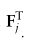
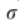
表示特征矩阵F中的第i和第j个全局原型,
是控制A的稀疏性的标准差参数。由于某些单元类型可能不包含在特定的域中,需要注意的是Fi也许 = 0。在这种情况下,我们将不再使用它进行进一步的计算,如2.4节所述。
2.2.3 Knowledge-aggregation-based预测
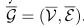
在该图中,顶点集V由V中的原始顶点(即全局原型)和查询单元的特征嵌入组成,得到一个扩展的特征矩阵
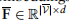
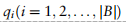
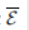
用新顶点的边展开。具体地说,通过添加全局原型和查询单元格之间的连接,得到了一个扩展的邻接矩阵A:
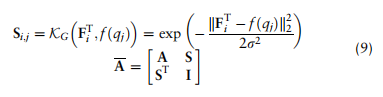
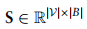
如下:

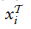
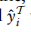
的靶细胞将被标记为”新型细胞类型”
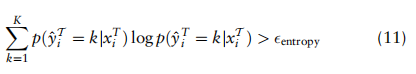
2.3具有类关系感知的数据集对齐
除了ZINB损失外,我们的模型在训练阶段还通过两种损失进行了优化,这有助于特征表示的域不变性和可区分性,如下节所述。
2.3.1关系对齐损失
这种损失的目的是调整不同数据集上的细胞类型的分布。在训练阶段,除了促进相同细胞类型特征的不变性外,还需要限制不同细胞类型的特征嵌入在潜在空间中的相对位置,特别是在从不同测序样本的不同测序平台收集的数据集时。基于这一思想,我们提出了由全局约束和局部约束组成的RAL:
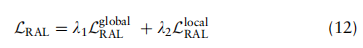
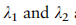
是折衷参数。对于全局项,我们促进了两个任意类之间的相关性在所有域上保持一致,这是通过测量A中块矩阵的相似性来实现的
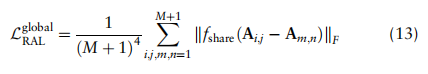
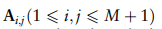
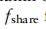
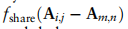
只保留行和列,其对应的全局原型为非零。在这种损失中,块矩阵的主对角元素的约束提高了块矩阵的类内不变性,而不同单元类型的关系相互依赖的约束促进了块矩阵其他元素相互依赖性的一致性。因此,优化这种损失有助于减少跨域的批处理效应。
三、结果
3.1竞争方法及评价指标
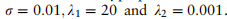
。在所有实验中,scMRA训练200次迭代,学习率为0.002。我们使用注释精度和调整后的rand索引作为评价索引。算法性能与注释精度的提高相关,因此更高的性能对应于注释精度。
3.2模拟研究
我们使用社区批准的R包飞溅来生成模拟数据(Zappia等人,2017)。我们考虑了许多在真实数据中可能存在的情况,例如源域的数量和单元格类型的数量。我们考虑了四种参考数据设置的情况来注释目标数据集,从一个源数据集到四个源数据集。我们还将不同细胞类型的数量从6到10不等。
对于上述20种组合中的每一种,我们通过将SplatSimulate函数的随机种子从1更改为10,生成了10个模拟数据集,从而得到20组模拟数据集。 dropout.-
type=’experiment’, dropout.shape =-1, dropout.mid= 0.5 and
de.facScale =0.2.
我们首先展示了在上述20组模拟数据集上,scMRA和其他四种竞争方法的性能的整体结果。对于每种方法和每一组数据集,我们计算出该方法在该组中所有10个不同数据集上的注释精度的平均值。我们用平均精度绘制了折线图,以观察这五种方法对域的数量和聚类的数量的敏感性(见图2)。一般来说,scMRA比不同的参数设置具有实质性的优势,特别是在多个参考数据集上。ItClust也提供了令人满意的结果,并表现优于Seurat和scmap。由于它未能学习低维表示和数据的内在识别,火星在模拟实验阶段获得了更差的性能。这种趋势是明显的,因为当聚类数量增加时,每种方法的精度都会降低,特别是对于Seurat和ItClust,但scMRA的精度降低得最小和最平稳。此外,随着参考数据的增加,除MARS外的所有方法均呈上升趋势。然而,在集群numbe的情况下
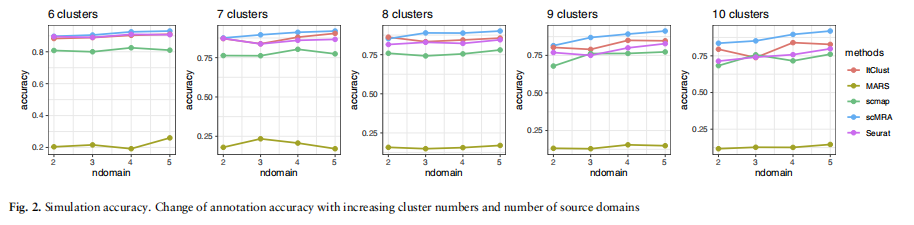
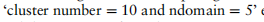
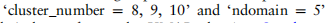
的模拟实验,并通过UMAP图可视化了它们的潜在特征(见补充图S1和S2)。我们可以很容易地看到,scMRA已经正确地注释了10个簇中的大多数细胞,只有scMRA可以清楚地分离出各种细胞类型,这表明了它在纠正批处理效应和注释未标记细胞方面的有效性。总的来说,scMRA在模拟数据中优于所有其他方法,尽管有集群的数量和参考数据集的数量。
3.3跨数据标注分析
我们首先从人类胰腺组织中选择了4个测序数据集进行实验:Baron_human(inDrop测序)(克莱因和马科斯科,2017)、Enge(智能-seq2)(皮切利等,2013)、穆拉罗(CEL-seq2)(哈希姆肖尼等,2012)和塞格斯托尔普(智能-seq2)。由于Baron_human中包含的细胞类型最为全面,我们将其设置为参考数据,同时将Enge、Muraro或塞格斯托尔普设置为目标。我们在补充表S1和表2中显示了使用scMRA的Enge、Muraro和serpe和四种不同竞争方法的注释结果。前者,也称为”接近情况”,使目标数据的标签空间与源数据的标签空间相同,而后者称为”部分情况”,目标数据的标签空间由源数据包含。
在表1中,对于接近设置的实验,我们可以看到scMRA在三个数据集上优于所有其他竞争方法,所有的准确率都超过93%。
表1。6组交叉数据实验中五种方法的注释精度值
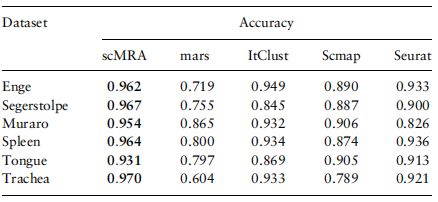
注:前三组以Baron_human作为参考数据,后三组以10个测序数据作为参考。表现最好的人用粗体显示。
让我们注意到表2,我们发现scMRA在部分情况下扩展了其优势,这实际上比接近的情况要复杂得多。
表2。在局部域自适应设置下,六组交叉数据实验中五种方法的注释精度值
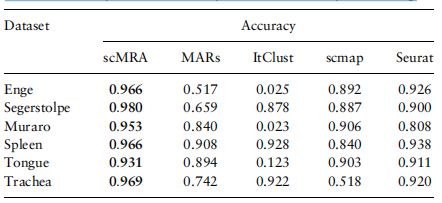
注:前三组以Baron_human作为参考数据,后三组以10个测序数据作为参考。表现最好的人用粗体显示。
在Enge和斯特格斯托尔普数据集中,scMRA的注释精度正在上升而不是下降,而在Muraro实验中仅下降了1%。而火星的性能,ItClustscmap和卫星下降同时比较关闭情况下,ItClust的注释精度的下降是最引人注目的,从94.9%到2.5%,从93.2%到2.3%,可能是因为ItClust没有使用目标数据的消息在训练阶段。为了可视化每种方法的性能,我们通过桑基图展示了部分情况下五种方法在Enge数据集上的注释精度。我们还通过UMAP将由scMRA、MARS和ItClust编码的潜在特征可视化,并将它们映射到一个共享的二维平面上(见补充图S3和S4)。
对于最大的细胞类型,”胰腺A细胞”(Enge中2282个细胞的998个细胞),在Sankey图中,我们可以看到scMSDA的注释准确率超过97%,而MARS仅达到50%.而ItClust很难将单个细胞与它真正的细胞类型配对。将我们的注意力转向最小的细胞类型”胰腺星状细胞”(Enge中2282个细胞中的998个细胞),scMSDA获得了高级别。相比之下,MARS、ItClust和scmap在注释来自该组的细胞时表现不佳。这表明scMSDA不仅能有效地对齐来自大群的细胞,而且还精确地注释了稀有细胞。至于UMAP图,我们可以看到,由于其强大的RAL,只有scMRA可以彻底分离所有六种细胞类型,没有明显的批效应。
们还选择了来自其他组织的测序数据来比较这些算法,包括脾脏、舌头和气管,都来自斑板项目(Schaumetal.,2018)。原作者分别用10个基因组学和Smart-seq2对这些组织进行了测序,并使用细胞本体论统一了注释的细胞类型。在这里,我们将10基因组学测量的数据作为参考数据,并以Smart-seq2测量的数据作为目标数据。scMRA在Smart-seq2脾脏、舌头和气管数据集上的注释准确率分别为96.4%、93.1%和97.0%,保持了其主导地位。此外,当将scMRA的注释任务从关闭域自适应设置转换为部分时,我们没有观察到scMRA性能的任何下降。本部分实验表明,抗参考数据集和目标数据集等多个数据集的干扰(特别是批效应)具有有效性、高泛化性能和强度。总的来说,这一证据充分证明了我们的算法适用于任何组织数据和测序技术(图3)。
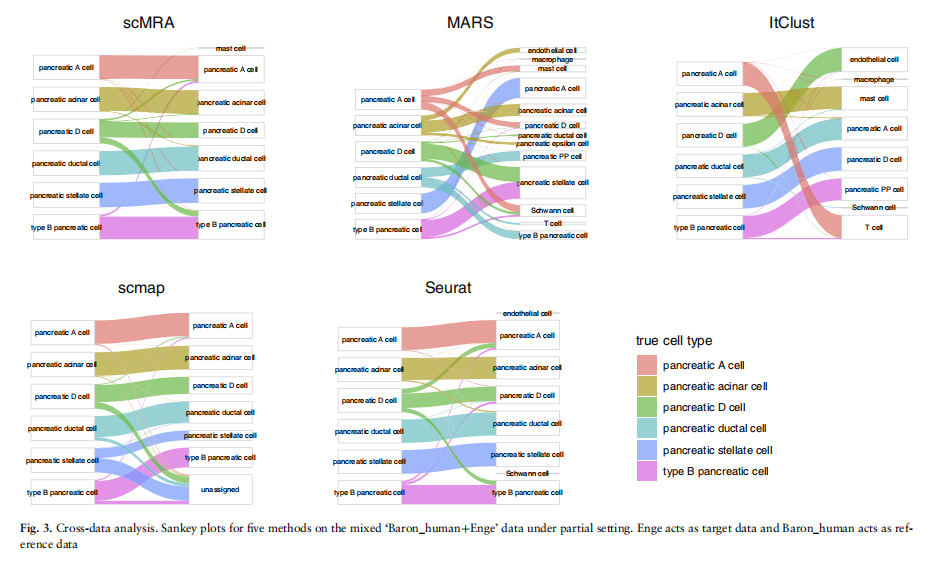
3.4多源数据标注分析
与其他注释方法相比,scMRA的另一个优点在于,它适合使用多个参考数据集对目标数据进行注释,即MSDA。当注释来自组织,甚至是不熟悉的物种的数据时,往往很难选择足够的参考数据。有时,引用数据在查询数据中不包含足够数量的单元格类型。其他时候,数据集可能太大,包含太多的不相关单元格,这可能会使分类复杂化。在这部分中,我们设计了两组实验来探索scMRA及其竞争方法面对多个参考数据集的性能,包括过多和不足的情况。前者意味着所有的引用数据集都比目标数据全面得多,后者表示任何单个引用数据集包含的单元格类型都太少,无法注释目标数据。
3.4.1过多的数据注释
我们从人类胰腺组织中选择了4个测序数据集进行实验:Baron_human(inDrop测序)、Enge(Smartseq2)、Lawlor(SMARTer测序)和Muraro(CEL-seq2)。由于Enge的单元格类型数量最少(总共6种),并且所有这些单元格类型都出现在所有其他三个数据集中,因此我们将其设置为目标数据。对于参考数据,我们依次使用Lawlor、LawlorþMuraro和LawlorþMuraroþBaron_human来观察给定越来越多的参考数据,五种方法的性能是否有上升趋势。我们在图4中显示了应用于上述三种情况的所有五种方法的注释精度值。左半部分显示了近域自适应设置下的结果,而右半部分显示了部分设置设置下的结果。首先,我们可以看到scMRA在所有5个注释准确率上都保持在95%以上。Seurat和ItClust跟随scMRA和MARS做得最差。对于除ItClust之外的三种竞争方法,在添加参考数据集时不能继续提高其性能。事实上,他在添加Baron_human作为参考数据时后退了一步,当穆拉罗加入劳勒时,scmap的准确性下降了
为了更好地更直观地展示我们的注释结果,我们使用UMAP来映射实验中由scMRA、MARS和ItClust编码的潜在特征,使用LawlorþMuraroþBaron_human作为参考数据,在部分设置为一个共享的二维平面下,并根据它们的真实细胞类型着色。将我们的注意力转向补充图S4,我们可以看到scMRA清晰地分离了六种细胞类型,而不受仅存在于参考数据中的细胞类型所带来的冗余信息的影响。与此同时,MARS,未能去除批处理效应,它将来自同一单元格类型的单元格对齐到不同的组中,导致注释精度较差。对于ItClust,像”胰腺腺泡细胞”这样的小群体不能与其他丰富的细胞类型区分开来,可能是因为它使用了比较距离与预测的细胞类型。
3.4.2数据注释不足
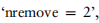
为例,我们从Lawlor中去除所有标记为1,2的细胞,从Baron_human中去除所有标记为3,4的细胞,从Muraro中去除所有标记为5,6的细胞。这些实验的详细信息见表3,以及所有五种方法的注释精度
表3。五种方法对三组数据不足实验的注释精度值
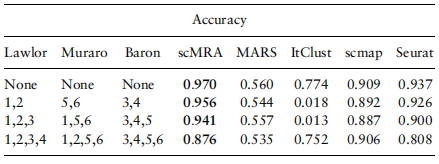
注意:左边的三列显示了在根据数据集中删除的单元格类型的索引。所有实验均以Enge作为目标数据,预处理的Lawlor、Muraro、Baron_human作为参考数据。表现最好的人用粗体显示。
我们可以看到scMRA相对于其他竞争方法的巨大优势。与它们在不删除任何细胞类型的情况下为Enge注释的显著性能相比,Seurat和scmap在每个参考数据集中包含的信息减少时遭遇了巨大的挫折。当我们从每个参考数据集中移除两个单元格类型时,ItClust突然崩溃,它的准确率仅下降到1.8%。然而,scMRA继续工作,注释精度几乎没有下降,即使每个参考数据集中目标数据中包含的六种单元类型中有四种被删除,这意味着参考数据和目标数据之间只有两种单元类型共享。更好地显示工作状态,删除批处理效果的能力和有效性标注未标记细胞最小scMRA信息,我们利用UMAP嵌入潜在特征提取的scMRA参考数据集通过删除2、3和4细胞类型每个到一个二维平面和彩色点的域他们属于(见补充图。S5).在所有三个实验中,我们看到scMRA完全混合了来自不同dna的细胞
3.5消融术的研究和分析
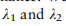
的适当间隔是相似的。
对于局部项,我们通过强制小批B中单元格的特征嵌入来接近其相应的全局原型,来增强每个类别的特征紧致性,从而得到以下损失函数:
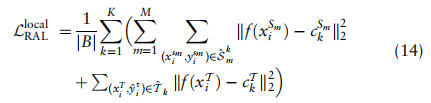
2.3.2分类损失
这组损失旨在提高特征的可区分性。基于对扩展知识图G中所有顶点的预测,将分类损失分别定义为全局原型、源单元和目标单元的三项组成:
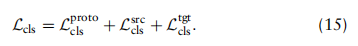
由于标签可用于全局原型和源单元,因此有两个交叉熵损失用于评估:
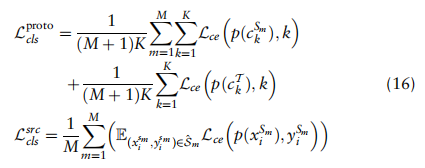

为交叉熵损失函数,p(x)表示x的分类概率。我们希望使他们的预测对目标细胞更具确定性,从而利用熵损失进行测量:
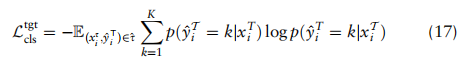
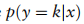
是x属于k类的概率。总体目标。结合上述定义的分类、ZINB和域自适应损失,基于ZINB模型的自动编码器fAE和GCN模型fG去噪的总体目标如下:
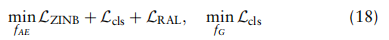
; 四、讨论
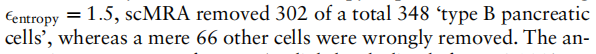
删除了总共348个”B型胰腺细胞”中的302个,而只有66个其他细胞被错误地删除。与以未经过滤的Baron_human作为参考数据的实验相比,scMRA的注释准确率从96.2%略有下降到94.0%。我们还将Muraro设置为目标数据,并在参考数据Baron_human中过滤出”B型胰腺细胞”。在Muraro中,scMRA将总共448个”B型胰腺细胞”中的392个注释为”新细胞类型”,只从其他细胞类型中删除了53个细胞。注释准确率为90.4%,仅比未过滤的情况低5.0%。这些结果表明,scMRA具有强大的能力
为了扩展我们的模型,我们还考虑了跨物种注释问题。我们选择了两个数据集,Baron_human和Baron_mouse(inDrop测序)进行实验。前者的数据集来自人类胰腺组织,而后者来自小鼠胰腺组织。尽管来自不同物种的细胞之间存在巨大差异,但Baron_human和Baron_mouse共享多达10种细胞类型。在Baron_human数据集中出现的14种细胞类型中,只有”肥大细胞”、”胰腺腺泡细胞”和”胰腺腺泡细胞”在Baron_human中是唯一的,而”B细胞”和”白细胞”在Baron_mouse中是唯一的。在过滤出Baron_human和Baron_mouse之间不共享的单元格类型后,我们以Baron_human作为参考数据查询Baron_mouse中的单元格。scMRA的注释准确率为83.3%,远高于MARS的58.5%和39.4%
对于ItClust。然后,我们将Baron_human作为查询数据,并将Baron_mouse设置为源数据,用从其他物种收集的参考数据来模拟来自人体组织的注释细胞。在这种情况下,scMRA的注释准确率上升到88.3%,而MARS的注释准确率达到63.9%,ItClust的注释准确率达到58.0%。虽然scMRA在跨物种注释任务中优于其他最先进的方法,但低于90%的准确率仍然表明scMRA在这方面有一些改进,以便在这方面具有真正的实用性。在未来,我们预计将进一步扩展我们的模型,以减少所有情况下错误分类细胞的比例。
5结论
在scRNA-seq分析中,细胞类型注释一直是一个重要的部分,尽管它是一个耗时和劳动密集型的部分。
在本文中,我们提出了一种多源域自适应方法scMRA来注释单细胞测序数据。scMRA假设一对细胞类型之间的关系在不同的数据集中是相似的,并将标签从多个参考数据集转移到目标数据,而不需要显式地纠正每个数据集中的批处理效应。通过用原型构造邻接矩阵并引入全局和局部RAL,我们的策略综合考虑了域级、簇级和单元级。我们的模型的这种结构决定了其对批处理效应干扰的完美适应性、平滑性和鲁棒性,特别适用于来自不同平台甚至组织的多个数据集的注释任务。
在仿真实验中,scMRA优于其他最先进的方法,包括MARS、ItClust、scmap和Seurat。scMRA的显著性能显示了其令人印象深刻的稳定性和鲁棒性的簇数和域数的巨大变化。实际数据从不同的测序平台,scMRA需要更大的领导和多个源数据实验模拟过多或不足参考数据来注释目标数据独立,scMRA删除批效应和识别细胞类型满意而没有其他最先进的方法相比能够维持他们的性能。
在实际应用中,目标数据的标签空间通常是由源数据包含在内的,所以我们主要关注所有实验中的部分案例。在部分设置下,注释算法通常会受到出现在参考数据中的单元格类型中的单元格的影响,但在目标数据中不存在。scMRA将这些额外细胞类型中的信息整合到邻接矩阵中,以更好地学习测序数据的嵌入。在实际数据实验中,我们发现,当其他所有方法从关闭设置切换到部分设置时,注释精度明显下降时,scMRA保持了优异的性能。甚至在相应的参考数据中查询恩格、塞格斯托尔和脾脏的性能也有所提高了。此外,在过量和不足的实验中,只有scMRA在部分设置的负面影响下保持其完整性,将细胞从不同的细胞类型中彻底分离,从不将细胞分配到唯一的参考数据细胞类型中。我们相信,与目前其他任何最先进的方法相比,针对部分设置和多个参考数据案例的定制为scMRA提供了更多的实用性和能力。
Original: https://blog.csdn.net/weixin_45156147/article/details/122887202
Author: Super齐
Title: scMRA:一种健壮的深度学习方法,可以用多个参考数据集注释scRNA-seq数据
原创文章受到原创版权保护。转载请注明出处:https://www.johngo689.com/697836/
转载文章受原作者版权保护。转载请注明原作者出处!