

目录
摘要
背景: 研究表明,rna 二级结构是由配对碱基构成的平面结构,在基本生命活动和复杂疾病中发挥着多种重要作用。Rna 二级结构图谱可以记录每个碱基是否与其他碱基配对。因此,准确预测二级结构有助于推断 rna 的二级结构和结合位点。通过生物实验和计算方法可以得到 rna 的二级结构图谱。其中,生物实验方法包括化学试剂法和生物结晶法。化学试剂法可以获得大量的预测数据,但成本高,且噪声大,由于测序覆盖范围有限,很难得到全部基因组 rna 的预测结果。相比之下,生物结晶法测定结果准确,但实验工作量大,成本高。另一方面,计算方法是交叉的,它包括一个三层完全连接的神经网络。然而,由于其网络结构较差,杂交不能完全了解 rna 二级结构特征,导致其性能较差。
结果
本文提出了一种基于双向LSTM和残差神经网络的新型端到端方法,用于预测RNA二级结构剖面。
结论
RPRes利用多种生物实验方法生成的数据集作为训练,验证和测试集来预测配置文件,这可以与许多预测要求兼容。与生物实验方法相比,RPRes降低了成本,提高了预测效率。与最先进的计算方法 CROSS 相比,RPRes 的性能显著提高。
背景
RNA通过转录,复制,蛋白质合成和基因表达调控在基础细胞过程和复杂疾病中起着各种重要作用[1-4]。它通常与多种蛋白质结合以参与细胞活动[5-7],其结构决定了其相互作用和功能[8,9]。RNA具有三层结构,包括一级结构,二级结构和三级结构[10]。其中,一级结构是基序,而二级结构是通过序列内自身折叠形成的平面结构,三级结构是通过二级结构元素的相互作用在空间中形成的。预测二级结构是鉴定三级结构的重要依据,也是了解各种生物活性RNA机制的重要前提[11]。不幸的是,预测不同长度的RNA的二级结构具有挑战性,并且大多数现有的计算方法都受到RNA长度的限制[12-14]。RNA二级结构曲线可以记录每个碱基是否与其他碱基配对。准确预测RNA二级结构剖面,不仅有助于推断出无长度限制的RNA二级结构[15],还有助于识别RNA的结合位点,从而促进RNA的功能研究。
RNA二级结构图谱可以通过生物学实验和计算方法获得。其中,生物实验方法涉及两种方式,即化学试剂和生物结晶。化学试剂法利用不同类型的探针来获得RNA二级结构剖面,根据探针类型可分为几类。RNA结构平行分析(PARS)[16-18]利用两种酶RNase V1(能够切割双链碱基)和S1(能够切割单链碱基)的催化活性来区分双链和单链碱基。通过引物延伸(SHAPE)分析的选择性2-羟基酰化采用高反应性化学探针,如1M6,NMIA(SHAPE)[19,20]和NAI-N3(icSHAPE)[21,22]来表征RNA谱。硫酸二甲酯(DMS)[23]使用小尺寸探针(CH3O)2SO2来表征RNA谱。虽然化学试剂法可以获得大量的预测数据,但其成本高且始终与高噪声相关,由于测序覆盖面有限,很难获得RNA上所有碱基的结果[24]。生物结晶方法可以从细胞中提取RNA进行结晶,然后通过使用核磁共振(NMR)或X射线晶体学获得RNA二级结构图[25]。生物结晶法可以获得准确的结果,但实验工作量大,成本高。在这方面,有必要采用计算方法来预测RNA二级结构剖面。现有的计算方法是CROSS [26],它创建了一个三层全连接的神经网络,并使用生物实验方法生成的数据作为训练,验证和测试集。在CROSS中,每个靶碱基(碱基将被预测到轮廓中)被处理成具有13个碱基的RNA序列,其中分别在靶碱基的前部和后部的6个。这些碱基在输入到 CROSS 之前,通过一热编码进行编码。因此,每条数据都被编码为 152 的向量。输入层有52个神经元,隐藏层有20个神经元,输出层有2个神经元。其相对较小的网络空间使其难以达到预期的性能。因此,有必要提出一种新的节省时间和人力的计算方法来预测RNA二级结构剖面。
结果
本文基于双向LSTM(Bi-LSTM)[27,28]和残差神经网络(ResNet)[29,30],提出了一种新的端到端预测方法”RPRes”。RPRes和CROSS既有相似之处,也有不同之处。相似之处在于RPRes和CROSS都基于神经网络,并且采用生物实验方法创建的多个数据集进行训练,验证和测试;通过这种方式,考虑了多个生物实验数据集的特征。不同之处在于,一方面,RPRes的网络空间大于CROSS的网络空间,因此它提取并学习了更多的特征;另一方面,RPRes的输入数据包含更多的上下文信息,以便更全面地了解每个目标基础。RPRes首先提取了基于Bi-LSTM层传输到相同格式输出的所有目标基础数据的特征,然后使用ResNet对Bi-LSTM的输出进行分类。在方法训练期间,每个数据集被随机分为三组:训练集(80%),验证集(10%)和测试集(10%)。训练集用于训练模型,验证集用于选择优秀模型,测试集用于测试模型性能。在方法测试和比较过程中,通过所有生物实验数据的测试集对RPRes的性能进行了测试和比较,并通过不同类型生物实验数据的测试集测试和比较了其泛化能力。
学习结果和演示
在本节中,介绍了RPRes的学习结果。将人类,小鼠,斑马鱼,PDB和酵母的数据集随机分为训练(80%),验证(10%)和测试(10%)集。将合并的训练集(包含多个生物实验数据集的所有训练集)和合并的验证集(包含多个生物实验数据集的所有验证集)用于30个epoch的训练和验证RPRes。图1表示实验中每个纪元的准确性和损失。可以看出,虽然在第10个时期,曲线由于梯度的急剧下降而波动,但合并验证集的整体精度逐渐增加后稳定,损失逐渐减少后稳定,表明RPRes可以成功预测RNA二级结构剖面。
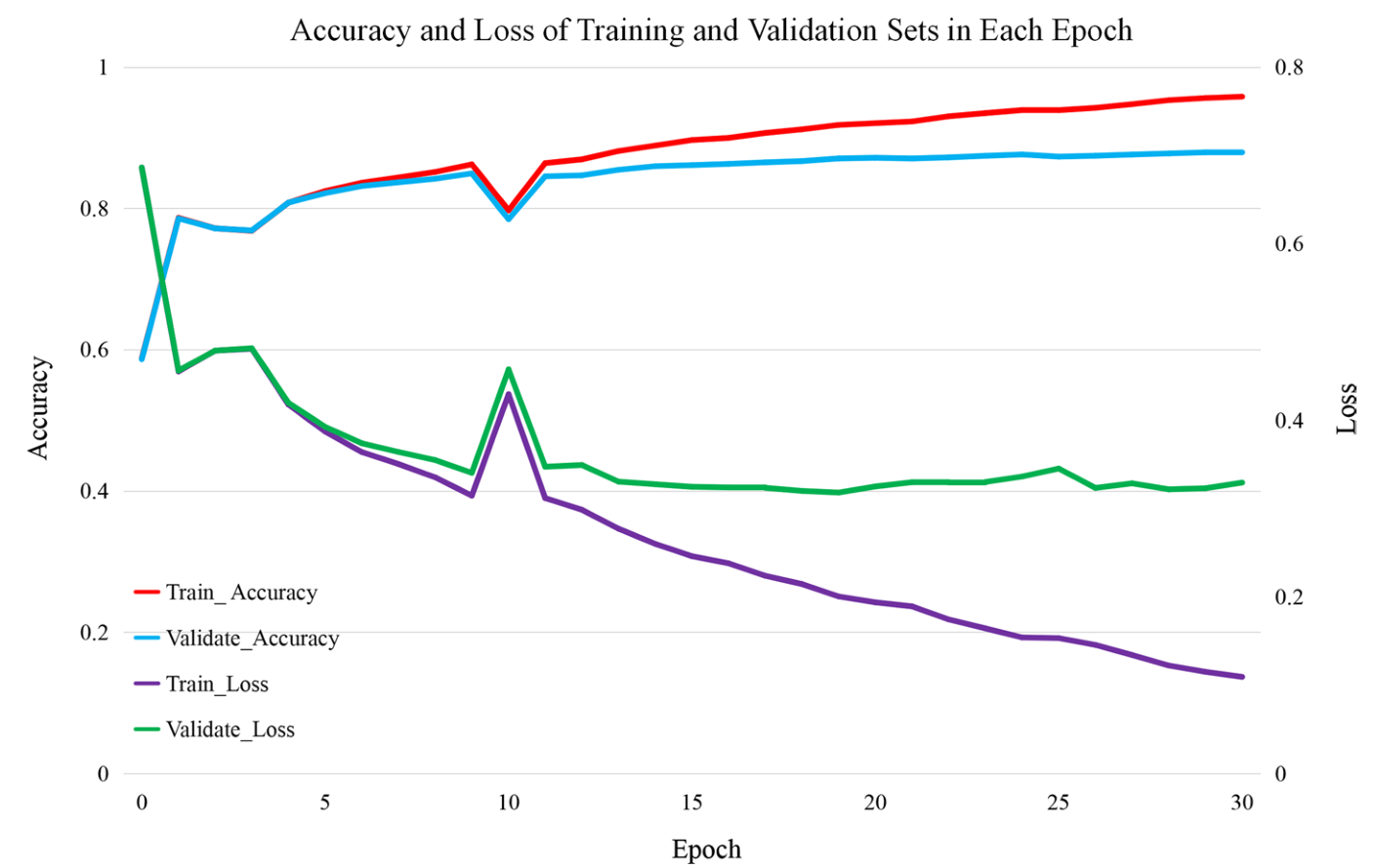

图1训练和验证集的准确性和损失,其中红色和蓝色曲线代表训练和验证集的准确性,而紫色和绿色曲线分别代表训练和验证集的损失值
预测结果和比较
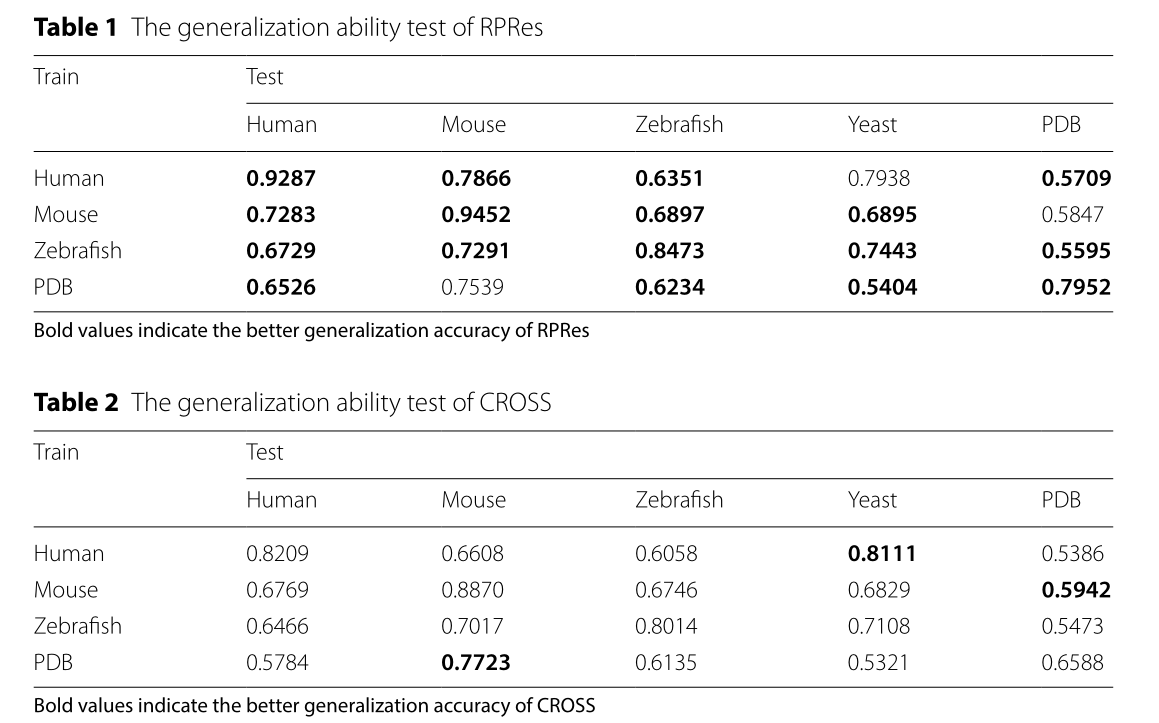
在本节中,介绍了RPRes的预测结果,并将其与CROSS的预测结果进行了比较。为了验证RPRes的性能,我们从两个方面将其与最先进的CROSS方法进行了比较,即合并测试集(包含多个生物实验数据集的所有测试集)中的性能和多个生物实验数据集的测试集之间的泛化能力。在第一个方面,人类,小鼠,斑马鱼,PDB和酵母的数据集被随机分为训练(80%),验证(10%)和测试(10%)集。将合并的训练、验证和测试集分别用于训练、验证和测试模型。采用准确度、灵敏度、精密度、F评分、MCC等指标[31]对RPRes和CROSS进行比较,绘制接收机工作特性(ROC)曲线,呈现两种方法的预测结果,并比较其曲线下面积(AUC)值。准确度定义为所有正确预测的目标碱基与总目标碱基的比率,灵敏度代表所有正确预测的双链碱基与总实际双链碱基的比例,精度是所有正确预测的双链碱基与总预测双链碱基的比例,F分数表示灵敏度和精度的加权谐波平均值, 而MCC是用于衡量二元分类的分类绩效的指数。它们相应的公式如下所示(Eqs. (见1-5),其中TP、TN、FP和FN分别表示真阳性、真阴性、假阳性和假阴性[32]。图2展示了 RPRE 和 CROSS 之间的性能比较。显然,就所有指数而言,RPRes取得了最佳性能。图3显示了 RPRes 和 CROSS 的 ROC 曲线。显然,RPRes 的 AUC 值大于 CROSS 的值。
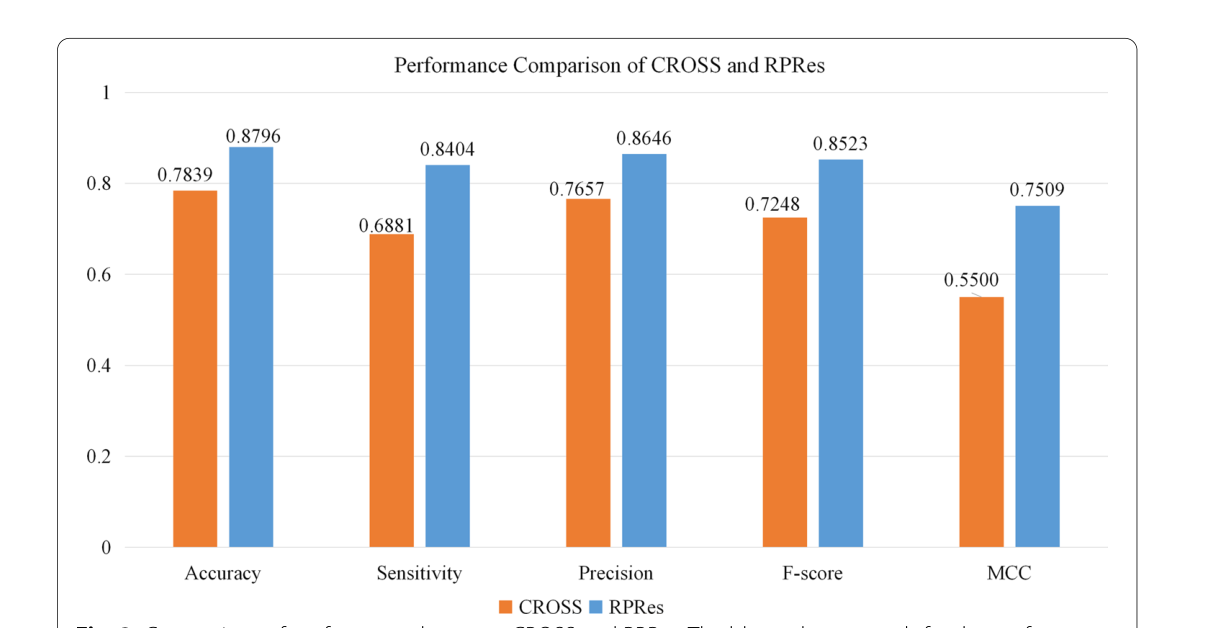
图2,cross和 rpres 的性能比较。蓝色列表示 rpres 的性能索引,橙色列表示 cross 的性能索引
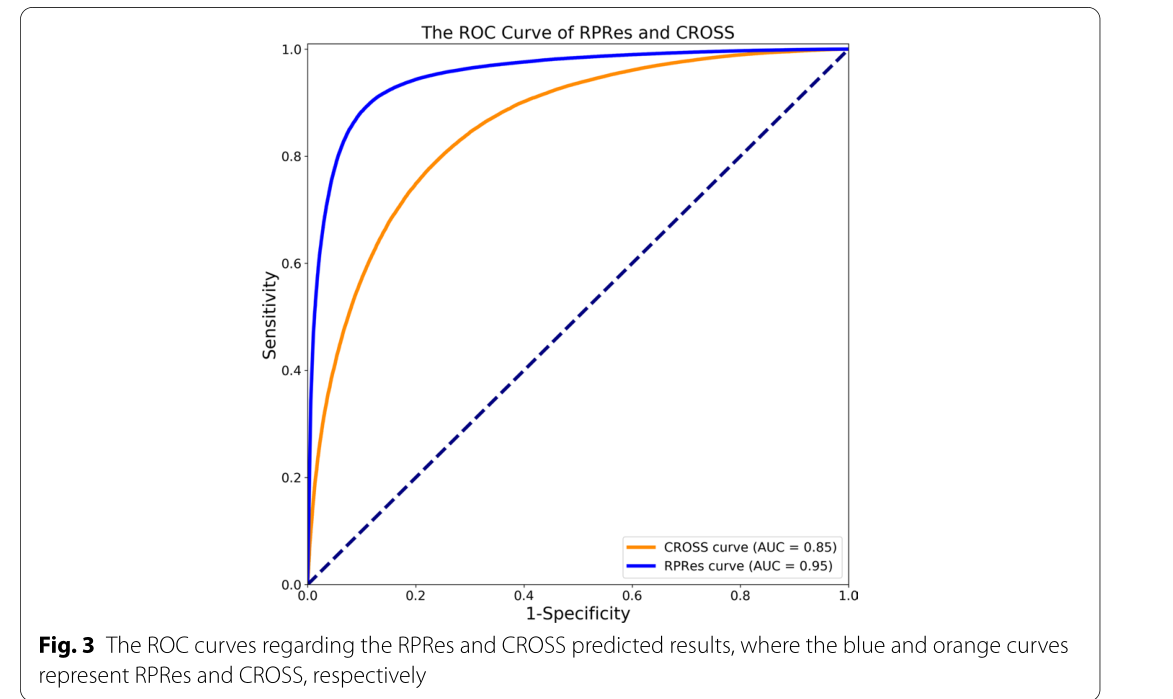
图3关于 rpres 和 cross 预测结果的 roc 曲线,其中蓝色和橙色曲线分别代表 rpres 和 cross
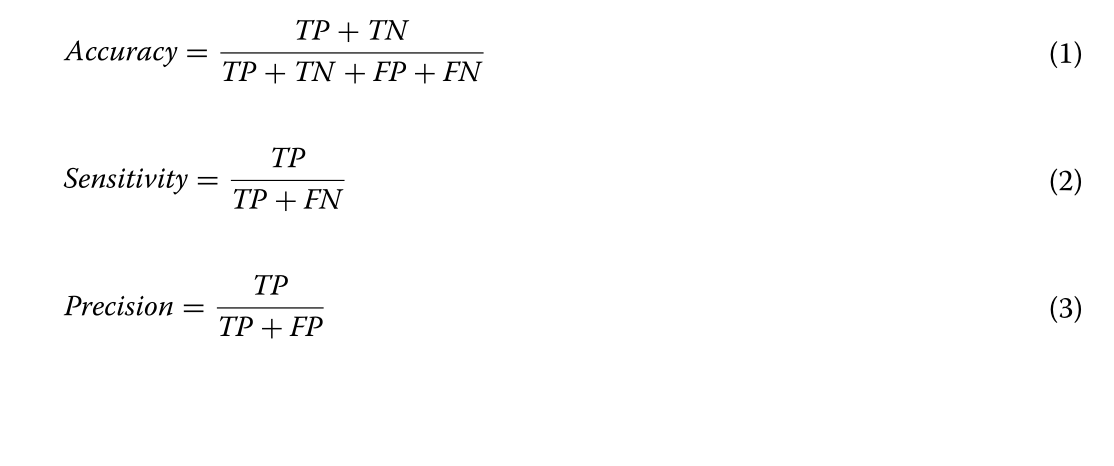
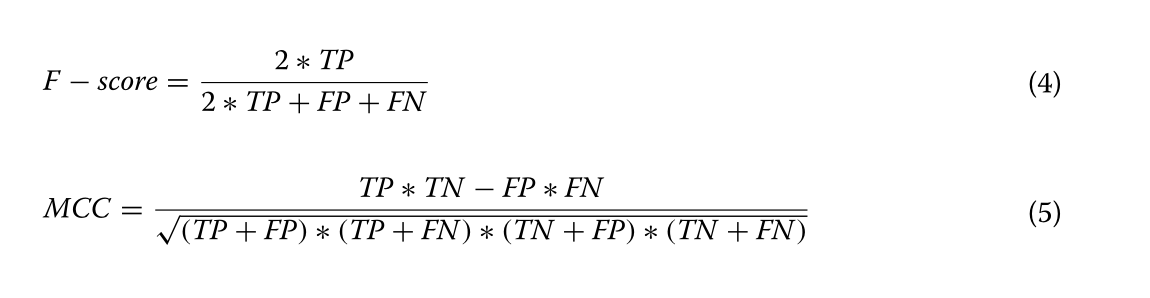
讨论
RNA是一种重要的生物大分子,参与几乎所有重要的生命活动和复杂疾病。RNA谱记录每个碱基是否与其他碱基配对,这有助于推断其二级结构和结合位点。传统的获取RNA图谱的生物实验方法费时费力,无法满足高通量数据的要求。此外,CROSS计算方法是一种三层全连接浅层神经网络,其相对较小的网络空间可能导致其性能较差。因此,迫切需要提出一种新的计算方法来完成剖面图预测。本文提出了一种基于Bi-LSTM和ResNet的新型端到端预测方法”RPRes”来预测RNA二级结构剖面。与生物实验方法相比,RPRes只需将数据输入算法模型即可获得预测结果,降低了成本,提高了预测效率。与CROSS相比,RPRes极大地改善了网络空间和输入数据上下文,更有效地提取和学习了目标基础的特征,从而提高了其性能。为了找到适当的上下文长度,我们截取了目标基地不同长度的上下文序列,包括149,119,89,59和29。图4显示训练 30 个 epoch 后不同长度上下文的准确性和损失。显然,不同长度上下文的精度接近,但损失值随着长度的增加而逐渐降低,表明过拟合概率较低,泛化能力较高。因此,我们在每个目标基地的前部切割了149个碱基,在每个目标基地的后部切割了149个碱基,并使用”N”来填充长度不足的序列。将每个靶标碱基加工成相同的格式,结果,RNA序列长度的多样性不影响RPRes。
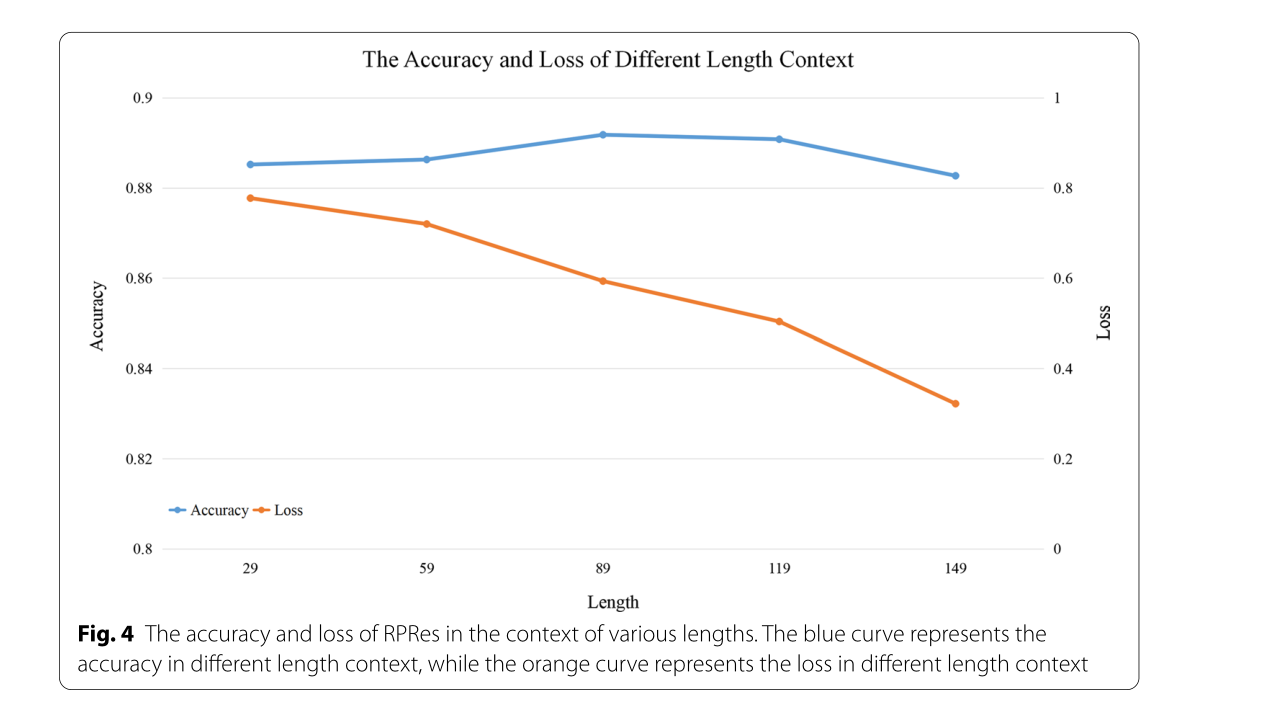
图4在不同长度的情况下 rpres 的准确性和损失。蓝色曲线表示不同长度上下文中的准确性,橙色曲线表示不同长度上下文中的损失
在RPRes的学习过程中,训练和验证集的准确性逐渐提高,而两组的损失逐渐减少。在过去的几个时期,验证集的损失和准确性趋于稳定,表明RPRes具有稳定的性能,并有效地预测了RNA二级结构剖面。在方法比较过程中,我们从两个方面比较了RPRes和CROSS。首先,我们比较了这两种方法的性能,使用相同的合并训练和验证集对其进行了训练和验证,然后比较了同一合并测试集的预测结果。具体而言,他们的表现是通过采用五个指标来衡量的:准确性,灵敏度,精度,F分数以及MCC和ROC曲线。从图2和3中观察到,RPRes将上述5个指数分别提升了12.20%、22.13%、12.91%、17.59%和36.52%,并将AUC值提升了11.76%。其次,我们通过对不同数据集的交叉测试,比较了两种方法的泛化能力。表1和2显示不同训练集的 RPRes 和 CROSS 的准确性。为了方便地显示每个交叉测试的性能的具体改进,表3显示了 RPREs 在各种交叉测试中的性能改进。显然,在大多数交叉测试中,RPRes的性能得到了改善,并且仅在人酵母,小鼠PDB和PDB-小鼠中降低了RPRes的性能。这种性能下降主要在化学试剂和生物结晶两种不同类型数据的交叉测试中观察到。主要原因是这两类数据的分布存在差异,CROSS的网络空间较小,导致对不同特征的学习较少,因此CROSS在这些交叉测试中的性能优于RPRes。评估神经网络模型的性能有很多方面,其中最重要的两个方面是同一测试集的性能和交叉测试集的泛化能力。RPRes在这两个方面优于CROSS,表明RPRes在预测RNA谱方面是有效且可接受的。有两个原因可能是导致 RPRes 到 CROSS 的卓越性能的主要原因。一方面,RPRes具有更大的神经网络空间,可以有效地学习和预测目标基础。另一方面,训练数据(299个碱基)比CROSS(13个碱基)长,后者包含更多特征。
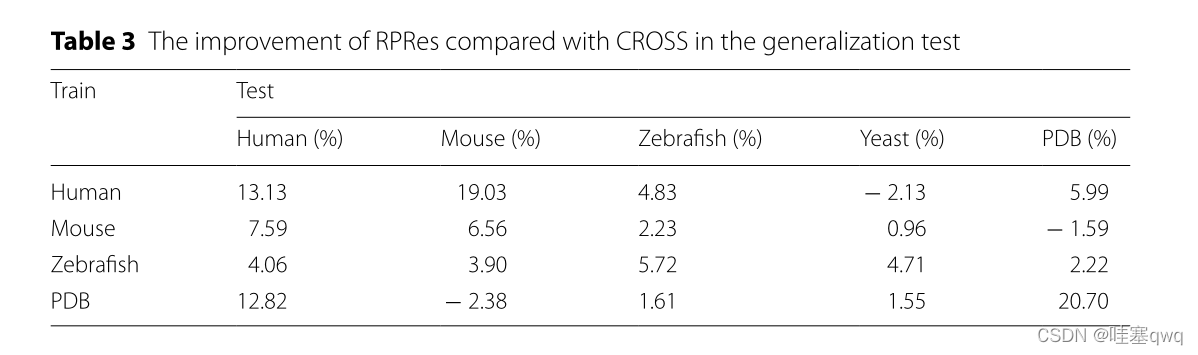
表3泛化试验中 rpres 与Cross相比的改进
结论
RPRes是一种基于深度学习的新型预测方法。该方法集成了各种神经网络技术,不同的技术在其运行中扮演着不同的角色。例如,Bi-LSTM可以提取输入数据的特征,并将这些数据集成到具有相同格式的输出中,从而便于通过下一层网络识别它们。ResNet将Bi-LSTM的输出分类为RNA谱。RPRes采用多样化的生物实验数据进行学习和训练,因此可以学习多个不同分布数据的特征;因此,该方法与许多预测要求兼容。同时,与生物实验方法相比,该方法大大降低了预测成本,与CROSS相比,其性能有所提高。因此,RPRes极大地有助于预测RNA二级结构特征和研究RNA功能。尽管RPRes在很多方面都很出色,但它仍然存在一些缺陷。在未来的工作中,RPRes的性能和泛化能力将得到进一步提高。同时,RNA谱预测是基础研究,可用于促进RNA二级结构和结合位点预测的研究。在未来的工作中,我们将在生物实验中应用RNA谱预测,从而引导生物学家获得更准确的结果,为生命科学研究做出贡献。
材料和方法
数据收集和处理
本文选取了多个生物实验数据集作为训练、验证和测试集。通常,化学试剂方法数据集来自两种技术,即PARS和icSHAPE,它们涵盖了斑马鱼[16],酵母[17],人类[21]和小鼠[21]全基因组数据。此外,从相关文献[25]中获得了生物结晶方法数据集(PDB),其中包含4667个RNA序列。由于原始数据不能直接用作RPRes的输入,因此有必要将这两种类型的数据处理成成熟的数据。
斑马鱼和酵母数据集是通过PARS技术获得的。该技术根据实验结果对RNA中的每个碱基进行评分,其中阳性值表示双链碱基,阴值表示单链碱基。我们根据评分对RNA中的每个碱基进行排序,并选择五个最低值作为单链靶碱基,而五个最大值作为双链靶碱基。根据斑马鱼的数据,筛选了53964个 rna 序列,包括557425个目标碱基,其中246853个双链碱基和310572个单链碱基。对于酵母数据,筛选了3196个RNA序列和34977个靶碱基,包括18095个双链碱基和16882个单链碱基。
通过染色质、核质和细胞质的 icshape 技术分别获得了人类和小鼠的数据集,并在体内和体外对其中的一些碱基进行了标记。双链碱基的得分接近0,单链碱基的得分接近1。在数据筛选过程中,我们分别筛选染色质、核质和细胞质中的靶基因。在人类数据中,我们选择了至少三个连续的可用得分作为候选基数。双链靶基因定义为体内和体外评分均为0的碱基,而单链靶基因定义为体内和体外评分均大于0.9的碱基。去除重复数据后,将这三个目标基组合为最终的人类目标基组。在小鼠数据中,我们还选择了至少三个连续的可用得分作为候选基数。然而,由于小鼠染色质、核质和细胞质之间的数据不一致,这三个数据集所选择的参数有一定的差异。在染色质和核质中,双链靶基因在体内和体外的连续碱基分别为0,而单链碱基分别在体内和体外的连续碱基分别大于0.9。在核质中,双链靶基因定义为体内和体外得分均为0的碱基,而单链碱基则为体内和体外得分均大于0.9的碱基。这三个目标基组,然后集成为最终的鼠目标碱基后删除重复的数据。从人类数据中筛选出6221个 rna 序列,包括399174个目标碱基,其中263334个双链碱基和135840个单链碱基。在小鼠数据中,筛选了11361个 rna 序列,包括778032个目标碱基,其中257969个双链碱基和520063个单链碱基。
从生物结晶方法获得的原始数据包含大量未知碱基和冗余序列。为了消除对RPRes性能的不利影响,去除了含有未知碱基的RNA序列,并利用CD-HIT [33]去除了同一性大于80%的冗余序列。最后,保留了502个RNA序列,其中包含225723靶碱基,包括94750个双链碱基和130973个单链碱基。
每个目标碱基的概况与其上下文序列密切相关。根据实验结果,我们将每个目标碱基的前部149个碱基和后部的149个碱基切割成一段数据,并用”N”填充不足的长度。通过这种方式,每个目标碱基被制作成一个具有相同长度的299个基地的数据。此外,这些碱基通过一热编码进行编码,因此每条数据都被编码为4*299矩阵。表4呈现一热编码的规则。

RPRes是一个全面的深度学习模型,包括Bi-LSTM [27,28]和ResNet [29,30]。RNA代表长距离上下文依赖性顺序数据,其轮廓与RNA上下文信息密切相关。有必要访问每个目标碱基的上下文特征以预测其轮廓。LSTM是一种特殊的递归神经网络,能够记录数据的远距离相关性信息。因此,选择Bi-LSTM作为模型中的第一层,提取目标碱基的上下文信息及其自身信息作为相同格式的输出,便于下一层网络的分类。RESNET是一种改进的深卷积神经网络,它利用捷径连接将输入数据和映射数据组合成输出数据,使网络的每一层都能包含真实的输入数据,有效地减少了因增加层数而导致的过拟合现象。因此,ResNet被选为Bi-LSTM输出的分类。图5显示模型的管线。在模型运行期间,当原始数据被处理成成熟数据(4∗299矩阵),它们被用作模型输入。利用一层Bi-LSTM将成熟数据编码为具有一致格式的输出,然后使用ResNet对Bi-LSTM编码数据进行分类。
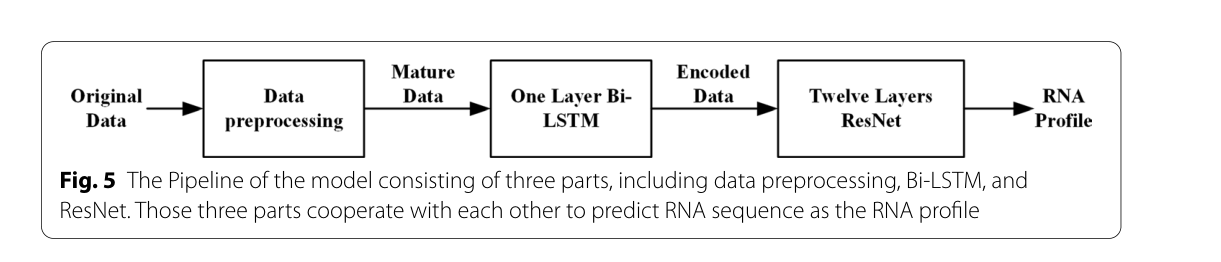
图5模型的流水线由三部分组成,包括数据预处理、双向LSTM和ResNet。这三个部分相互配合,预测RNA序列作为RNA剖面
Bi-LSTM _:_递归神经网络(RNN)已广泛应用于文本,音频,视频和其他顺序数据的研究[34,35]。当采用RNN处理顺序数据时,给定时刻神经元的输出作为下一时刻神经元的输入;因此,RNN 可以有效地利用上下文信息。然而,传统的RNN具有有限的记忆能力,随着序列长度的增加,这将失去在上下文中学习信息的能力,使其容易陷入梯度消失。LSTM是一种特殊类型的RNN,可以通过引入栅极机制来解决梯度消失的问题;因此,它在表示上下文信息和从顺序数据中提取长距离依赖性特征方面优于传统的RNN。LSTM 中有三个门,包括输入门、忘记门和输出门。输入门决定在单元状态下存储的新信息,忘记门决定要丢弃的信息离开单元状态,而输出门根据单元状态决定要用作输出的信息。图6是LSTM单元的图,可以实现如下(Eqs. 6–10):
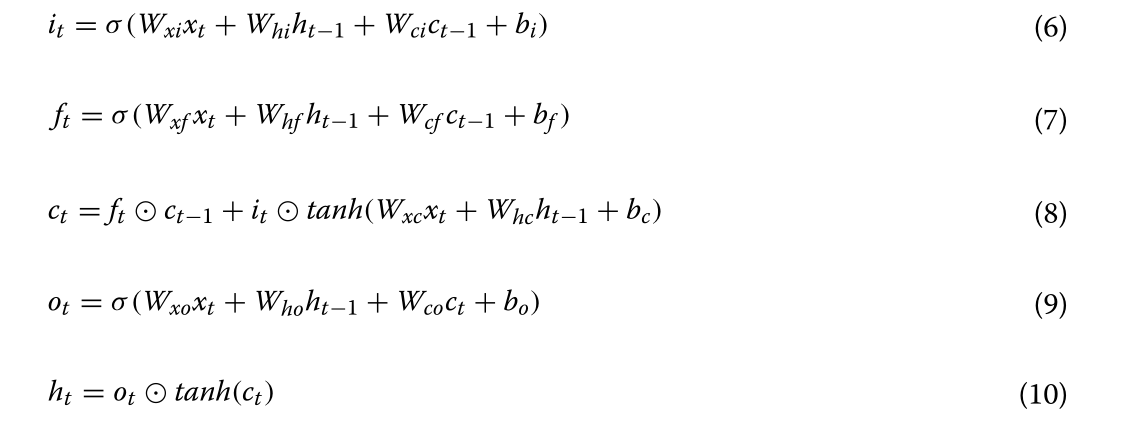
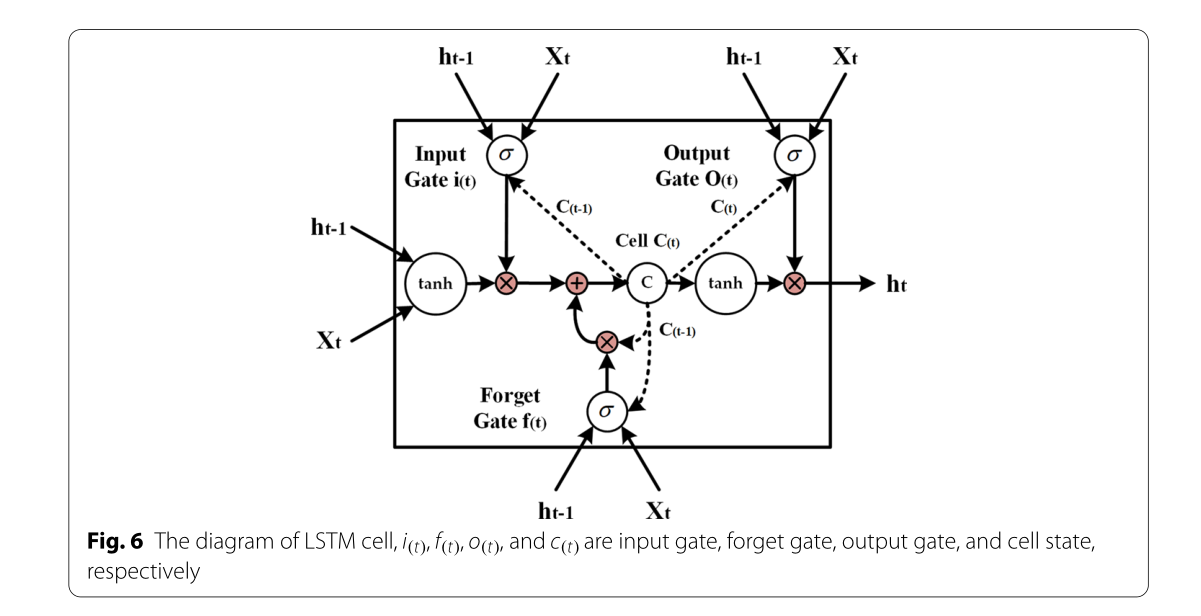
其中,σ表示逻辑Sigmoid函数,而i、f、o和c分别表示输入门、遗忘门、输出门和单元矢量,并且所有这些因子与隐藏矢量h具有相同的维度。同时,w表示权重矩阵,而b表示偏置矢量。
本文利用单层Bi-LSTM提取了由前向和后向网络组成的所有目标碱基数据的上下文特征。前向 LSTM(512 个隐藏节点)从左到右处理目标序列,而后向 LSTM(512 个隐藏节点)按相反顺序处理目标序列。因此,获得了两个隐藏状态序列,其中一个来自前向网络,另一个来自后向网络。此外,Bi-LSTM连接了每个基站的前后隐藏状态,目标碱基的串联状态(1024个节点)为输出。图7显示了 Bi-LSTM 的示意图。
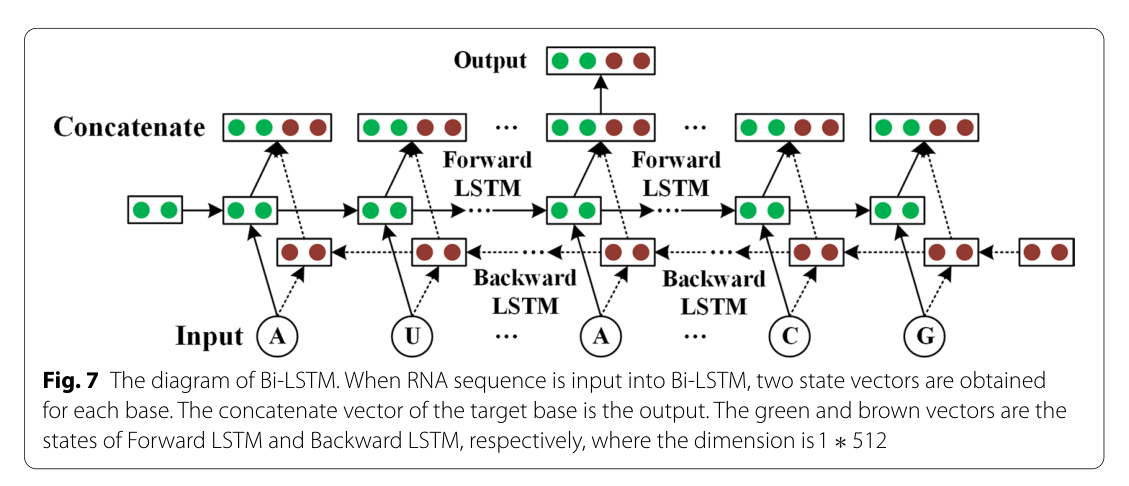
图7 Bi-LSTM的示意图。将RNA序列输入Bi-LSTM,得到每个碱基的两个状态向量。目标基数的串联向量就是输出。绿色和棕色向量分别是前向LSTM和后向LSTM的状态,其中维度为1∗512
RESNET:深卷积神经网络[36]已被广泛用于二维数据的识别和分类。事实证明,深卷积神经网络的深度是至关重要的,它有助于丰富特征,提高精度[37]。然而,卷积神经网络的精度并不总是随着深度的增加而提高,相反,当精度达到饱和时,其精度可能会下降。当卷积神经网络达到饱和时,为了在增加新的层的情况下保持其饱和精度,新的层必须是一个单位映射层:H(X)=x,这将导致深层网络退化为浅层网络。不幸的是,随着层的增加,可能会遇到梯度消失或爆炸[38],使得新层很难适应身份映射。因此,单纯提高神经网络的深度并不能满足研究的需要。RESNET通过每个残差块[29]的输入卷积层和输出卷积层之间的捷径连接有效地解决了这个问题。一般来说,ResNet包含一定数量的残留块,它们是ResNet模型的核心组件。图8[29]显示了残留块的示意图。在残差块中,输入表示为x,残差块的映射为F(X),输出H(X)=F(X)+x,在ResNet中,当网络达到饱和时,F(X)学习等于0,因此残差块成为单位图,这比普通卷积网络中的学习H(X)=x容易得多[29]。在反向传播过程中,H(X)的偏导数如下式(式)所示。11)。
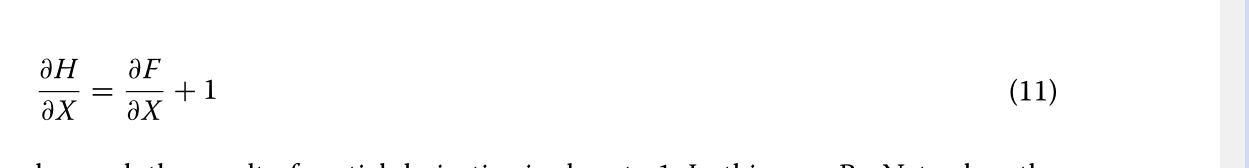
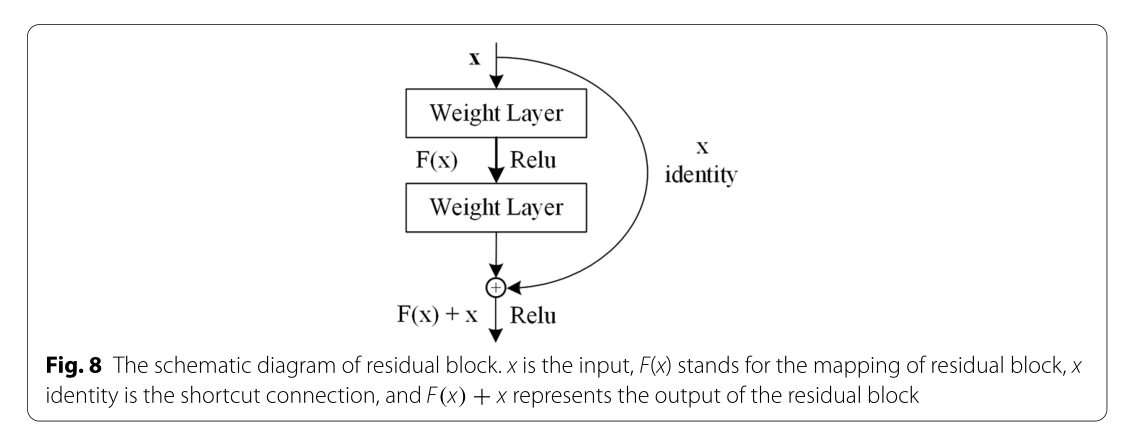
图8残差块示意图。X表示输入,F(X)表示残差块的映射,x标识表示快捷连接,F(X)+x表示残差块的输出
在本文中,ResNet涉及三种类型的残差块,其内核相同3∗3,内核的维数分别为 16、32 和 64。图9显示 ResNet 的原理图。从图中可以看出,输出(1∗1024)的 Bi-LSTM 首先被转换为二维数据 (32∗32),然后用作 ResNet 的输入数据。在 ResNet 中,具有 16 维的卷积神经网络3∗3卷积核优先计算Bi-LSTM的输出数据,输出包含16维32∗32数据。其次,使用残余块来计算输出。这些残余块的卷积核的维数分别为16、16、32、32、64和64。因此,残余块的输出是 64 维32∗32数据。此后,采用全局平均池化层将残差块的输出数据汇集到1∗64向量。最后,采用全连接层将残留块的输出分类为RNA二级结构剖面。
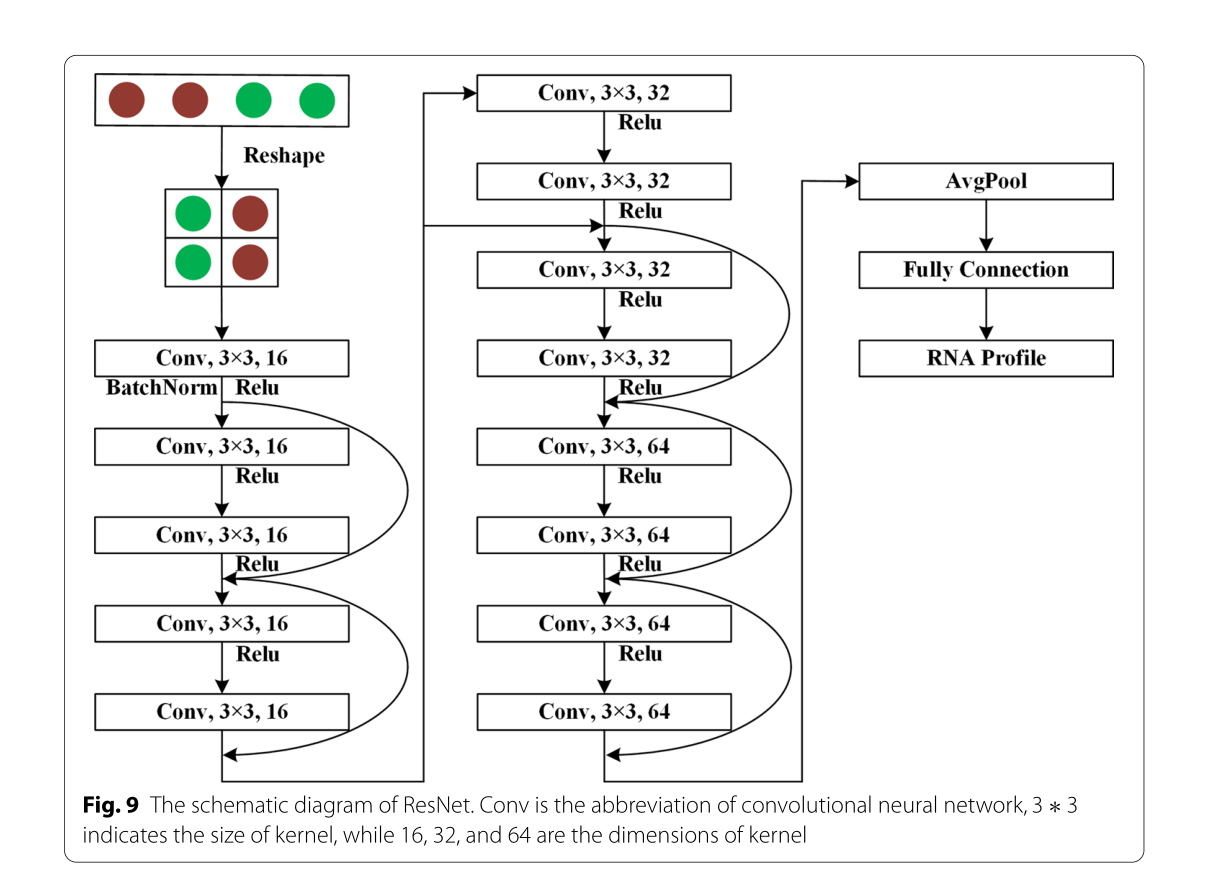
图9 ResNet原理图。conv是卷积神经网络的缩写,3 *3表示核的大小,16、32、64表示核的维度
Original: https://blog.csdn.net/qq_52914392/article/details/123140344
Author: 哇塞qwq
Title: 基于双向 lstm 和残差神经网络的 rna 二级结构预测方法
原创文章受到原创版权保护。转载请注明出处:https://www.johngo689.com/690852/
转载文章受原作者版权保护。转载请注明原作者出处!