

SUM1 读论文
- MGraphDTA: deep multiscale graph neural network for explainable drug–target binding affinity prediction
* - 1. background
- 2.模型总览
- 3. 模型原理
–- 3.1 输入
- 3.2 图神经网络
+ - 3.2.1 Message passing phase
- 3.2.2 Readout phase
- 3.3 多尺度图神经网络用于药物编码(encoding)MGNN
+ - 3.3.1 多尺度块
- 3.3.2 Transition layer
- 3.4 Multiscale convolutional nerual network for target encoding
- 3.5 MGraphDTA network architecture
- 3.6 gradient-weighted affinity activation mapping
- 3.7 数据集(待补充)
- 4. 实验结果(待补充)
+ - 4.1 分类任务
- 4.2 回归任务
- 4.3 在更真实条件下的效果评估
- 4.4 模型简化测试
- 4.5 视觉解释性
- 4.6 MGNN如何克服过平滑问题
- 4.7 局限性
- 5 结论
论文名称:MGraphDTA: deep multiscale graph neural network for explainable drug–target binding affinity prediction
论文下载地址:
MGraphDTA
论文代码下载地址:
MGraphDTA and Grad-AAM
数据集下载地址:
Davis and KIBA filtered Davis Metz 、Tox-Cast Human and C. elegans datasets
MGraphDTA: deep multiscale graph neural network for explainable drug–target binding affinity prediction
图神经网络广泛应用于预测靶向药物亲和力(Predicting drug–target affinity (DTA)),然而目前的浅层图神经网络不足以捕获化合物的全局特征。
1. background
对于预测靶向药物亲和力的模型一般可分为三类:
-
基于结构的模型:考虑小分子和蛋白质的3D结构.
-
基于特征的模型:代表方法是蛋白质化学计量学(proteochemometrics),依赖蛋白质和配体的清晰描述:生物特征向量,用一组确定的二进制数表示药物是否作用于目标。得到的生物特征向量可以用于训练深度学习模型。如前馈神经网络(FNN)、支持向量机(SVM)、随机森林(RF)以及其他的核(kernel)模型。其中FNNs的效果最好。
- 基于深度学习的模型:深度学习模型源于新兴技术如high throughput screening, parallel synthesis采集到的生物医药与复合物活性数据。如DeepDTA用两层卷积神经网络(CNN)分别学习药物和蛋白质的结构表达,之后利用多层感知机预测药物匹配关系。WideDTA在此基础上改进了输入(增加两个额外的文本输入),总计四个输入。总之,基于CNN的模型被证明是有效的,并且基于CNN识别代表化合物结构的二维数据是被证明可行。 之后对于CNN不能很好的表达分子结构的信息的缺点,引出了图神经网络(GNNs),GNNs用图来表示药物,并用GNN(频域方法?)实现DTA预测。经验证,基于GNNs的模型要优于CNNs 。
如GraphDTA对比了如图卷积网络(GCN)、图注意力网路(GAT)、GIN(?)和GAT-GCN用于DTA预测(回归问题)。注意力机制的引入增加了模型 的可解释性。
另一方面,使用蛋白质的结构相关信息(structural-related features of protein)作为输入以提升DTA预测也被证明可行。(个人感觉该方法类似将先验知识融入深度学习算法),如DGraphDTA将蛋白质序列作为输入,但由于蛋白质结构信息的不全可知,所以利用预测的contact map作为输入。
总之,目前的浅层GNNs有三种问题: 1.浅层结构无法很好的捕捉全局特性。 因此,需要多层堆叠的图卷积层,若想捕获K跳邻居的结构,应该堆叠k个图卷积层(不太理解,这里是指embedding of nodes?deep encoder 有点相似,以后查证)


2.GNN应保留局部信息。
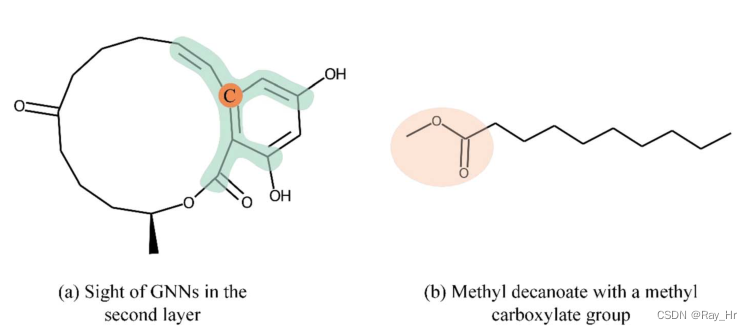
3.基于图形的DTA可解释性依赖于注意力机制。 (GAT网络?)注意力机制有较好的数学解释,但仅考虑顶点领域。

; 2.模型总览
为了解决上述问题,该文章提出了多尺度图神经网络multiscale graph neural network (MGNN) 以及可视化模型:权重梯度相关性图gradient-weighted affinity activation mapping(Grad-AAM) 。
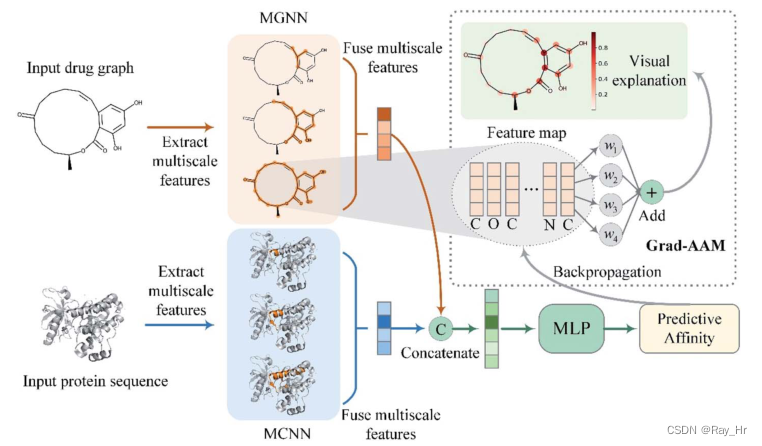
预测部分:MGNN用来27层图卷积层以及多尺度卷积神经网络(MCNN)用于提取药物和靶点的多尺度特征,融合后获得对于关系,输入多层感知机以预测亲和力。
可视化部分:Grad-AAM根据MGNN的最后一层输入的梯度以生成概率图模型,用以标注对DTA最有贡献的原子。Grad-AAM源于在图像中梯度加权类激活映射(gradient-weighted class activation mapping (Grad-CAM) ),对其CNN的部分替换为GNN。(最近好多模型都是将图像领域的CNN结构替换为GNN)。
3. 模型原理
3.1 输入
模型输入是j简化分子线性输入系统SMILES(Simplified Molecular Input Line Entry System),用短ASCII码来表示化学结构,输入的目标是一组蛋白质序列(字符串),其中每一个字符代表一个氨基酸。(氨基酸是蛋白质的基本结构?不太了解)。利用RDKit将SMILES处理成具有点或边的图结构以及邻接矩阵。
对于蛋白质序列,本文首次建立了一个词汇表将每一个字符映射成整型(int),如下所示:
氨基酸右对齐丙氨酸1半胱氨酸2谷氨酸4
为了节省训练成本,将蛋白质序列长度限制在1200,基本上至少覆盖了80%的蛋白质,之后经过embedding 层将器转化为128维向量(embedding space)one-hot 编码虽然也可encode蛋白质,但是不能描述两个不同氨基酸之间的语义相关性,例如不同的one-hot向量都具有零余弦相似性。(看到one-hot总想到交叉熵损失,1001和0110就存在零余弦相似性,不知是否有解决的方法,回头查一查)
3.2 图神经网络
图可以被表示为:
G = ( ν , E ) \mathcal{G}=(\nu, \mathcal{E})G =(ν,E )
v i ∈ ν v_{i} \in \nu v i ∈ν代表第i个原子,e i j ∈ E e_{i j} \in \mathcal{E}e ij ∈E代表了第 i 和第 j 个原子连接的边。GNN通常利用message pass 和 readout将图G {G}G映射到向量空间y G ∈ R d y_{\mathcal{G}} \in \mathbb{R}^{d}y G ∈R d,其中message pass根据领域更新顶点信息,readout为整张图计算特征向量。
3.2.1 Message passing phase
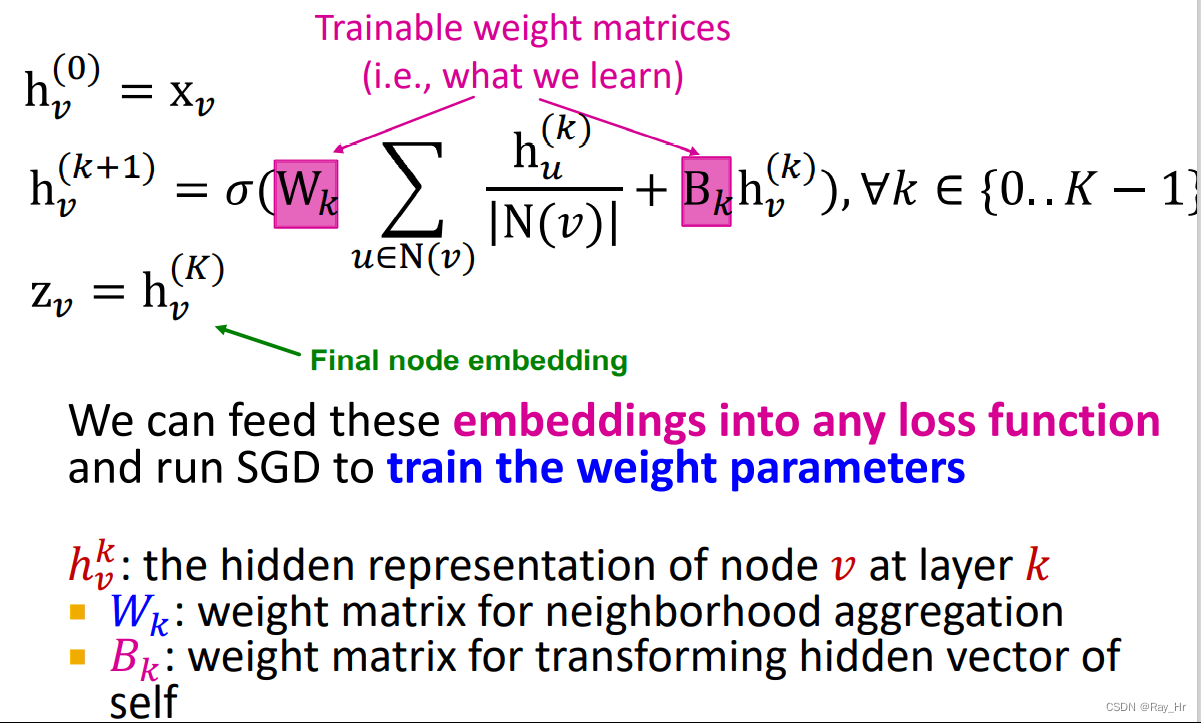
用如下的图卷积层来实现随着时间步长t的变化第i个顶点的embedding 向量的更新
x i ( t + 1 ) = σ ( W 1 x i ( t ) + W 2 ∑ j ∈ N ( i ) x i ( t ) ) x_{i}^{(t+1)}=\sigma\left(W_{1} x_{i}^{(t)}+W_{2} \sum_{j \in \mathcal{N}(i)} x_{i}^{(t)}\right)x i (t +1 )=σ(W 1 x i (t )+W 2 ∑j ∈N (i )x i (t ))
上式中W 1 W_{1}W 1 和W 2 W_{2}W 2 是可学习的权重矩阵,N ( i ) \mathcal{N}(i)N (i )是顶点i的邻居(一阶邻居?),σ \sigma σ就是批标准化和激活函数,本文中选择ReLU。由上式可知,通过不断的迭代可以逐渐捕获图上全局信息。(该方法类似于GCN,如下所示,不知是否可以引入图注意力网络GAT,通过调参来实现更好的预测结果,或者使用较新的异质图模型)
; 3.2.2 Readout phase
就是对最后一层输出的顶点embedding vector取均值:
y G = 1 ∣ V ∣ ∑ v ∈ V x v ( L ) y_{\mathcal{G}}=\frac{1}{|\mathcal{V}|} \sum_{v \in \mathcal{V}} x_{v}{ }^{(L)}y G =∣V ∣1 ∑v ∈V x v (L )
∣ V ∣ |{V}|∣V ∣代表分子图中顶点的数量,L是最后一步时间步。readout层将节点的embeddings 聚合到图的 embedding。(应该是用于后文的可视化,该点可能是本文的创新处之一)
3.3 多尺度图神经网络用于药物编码(encoding)MGNN
Intuition: chemical
MGNN包括了三层多尺度块并通过transition层连接。
3.3.1 多尺度块
多尺度块源于DenseNet,本文将dense connection 迁移到了GNN.
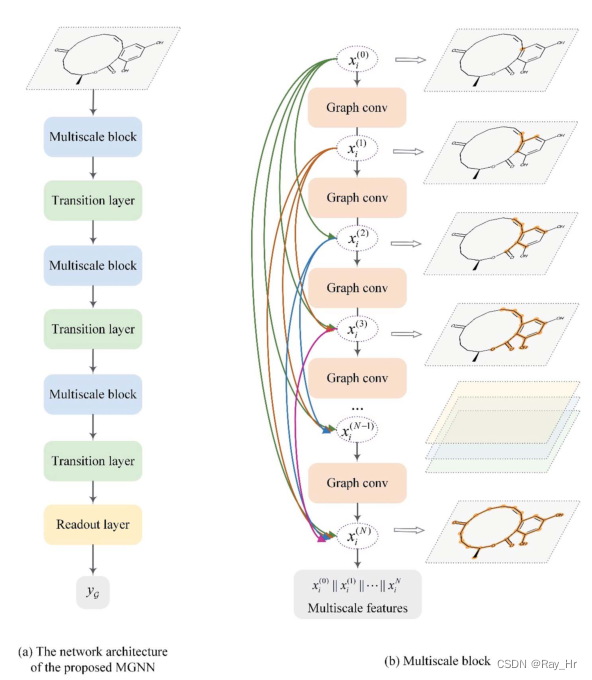
dense connection 将每一层和其他层通过前向通道连接起来,这样所有层都可以直接接受关于每个权重的损失函数的梯度,这样梯度就不会在层与层之间传递而引起梯度消失,因此该方法可以将网络拓展到更深的层次(很牛,有点感觉像强化版resnet)。多尺度层可以用如下表示:
x i ( 1 ) = H ( x i ( 0 ) , Θ 1 ) x_{i}^{(1)}=\mathscr{H}\left(x_{i}^{(0)}, \Theta_{1}\right)x i (1 )=H (x i (0 ),Θ1 )
x i ( 2 ) = H ( x i ( 0 ) ∥ x i ( 1 ) , Θ 2 ) x_{i}^{(2)}=\mathscr{H}\left(x_{i}^{(0)} \| x_{i}^{(1)}, \Theta_{2}\right)x i (2 )=H (x i (0 )∥x i (1 ),Θ2 )
x i ( 3 ) = H ( x i ( 0 ) ∥ x i ( 1 ) ∥ x i ( 2 ) , Θ 3 ) x_{i}^{(3)}=\mathscr{H}\left(x_{i}^{(0)}\left\|x_{i}^{(1)}\right\| x_{i}^{(2)}, \Theta_{3}\right)x i (3 )=H (x i (0 )x i (1 )x i (2 ),Θ3 )
x i ( N ) = H ( x i ( 0 ) ∥ x i ( 1 ) ∥ ⋯ ∥ x i N − 1 , Θ N ) x_{i}^{(N)}=\mathscr{H}\left(x_{i}^{(0)}\left\|x_{i}^{(1)}\right\| \cdots \| x_{i}^{N-1}, \Theta_{N}\right)x i (N )=H (x i (0 )x i (1 )⋯∥x i N −1 ,ΘN )
H \mathscr{H}H就是上文提到的卷积层,Θ n \Theta_{n}Θn 就是n层的参数(W 1 、 W 2 W_1、W_2 W 1 、W 2 ),∥ \|∥是串联操作,多尺度块因此提取到多尺度信息(感觉这里的多尺度就是指节点的n跳领域),这些信息在局部和全局中描述了分子的结构信息
; 3.3.2 Transition layer
为了增加MGNN的网络深度,将两块多尺度块用transition层连接起来。连接层的作用一是集成上一个层多尺度块所获取的多尺度特征,二是减少特征图通道数。对于第N+1时间步的多尺度信息x i ( 0 ) ∥ x i ( 1 ) ∥ ⋯ ∥ x i N ∈ R d + ( N − 1 ) h x_{i}^{(0)}\left\|x_{i}^{(1)}\right\| \cdots \| x_{i}{ }^{N} \in \mathbb{R}^{d+(N-1) h}x i (0 )x i (1 )⋯∥x i N ∈R d +(N −1 )h多,transition层表示如下:
x i ( N + 1 ) = σ ( Φ 1 ( x i ( 0 ) ∥ x i ( 1 ) ∥ ⋯ ∥ x i N ) + Φ 2 ∑ j ∈ N ( i ) ( x j ( 0 ) ∥ x j ( 1 ) ∥ ⋯ ∥ x j N ) ) \begin{aligned} x_{i}^{(N+1)}=& \sigma\left(\Phi_{1}\left(x_{i}^{(0)}\left\|x_{i}^{(1)}\right\| \cdots \| x_{i}^{N}\right)\right.&\left.+\Phi_{2} \sum_{j \in \mathcal{N}(i)}\left(x_{j}^{(0)}\left\|x_{j}^{(1)}\right\| \cdots \| x_{j}{ }^{N}\right)\right) \end{aligned}x i (N +1 )=σ(Φ1 (x i (0 )x i (1 )⋯∥x i N )+Φ2 j ∈N (i )∑(x j (0 )x j (1 )⋯∥x j N )
Φ 1 , Φ 2 ∈ R ( M / 2 ) × M \Phi_{1}, \quad \Phi_{2} \in \mathbb{R}^{(M / 2) \times M}Φ1 ,Φ2 ∈R (M /2 )×M是可学习的权重矩阵,M = d + ( N − 1 ) h M=d+(N-1) h M =d +(N −1 )h(N-1为时间步,d为特征,h为顶点数)通过Φ 1 , Φ 2 \Phi_{1}, \quad \Phi_{2}Φ1 ,Φ2 的维度可以看出,经过transition层后通道回转为原来的一半,从而节省计算时间。最后readout层就可用来将整张图转化为特征向量。
3.4 Multiscale convolutional nerual network for target encoding
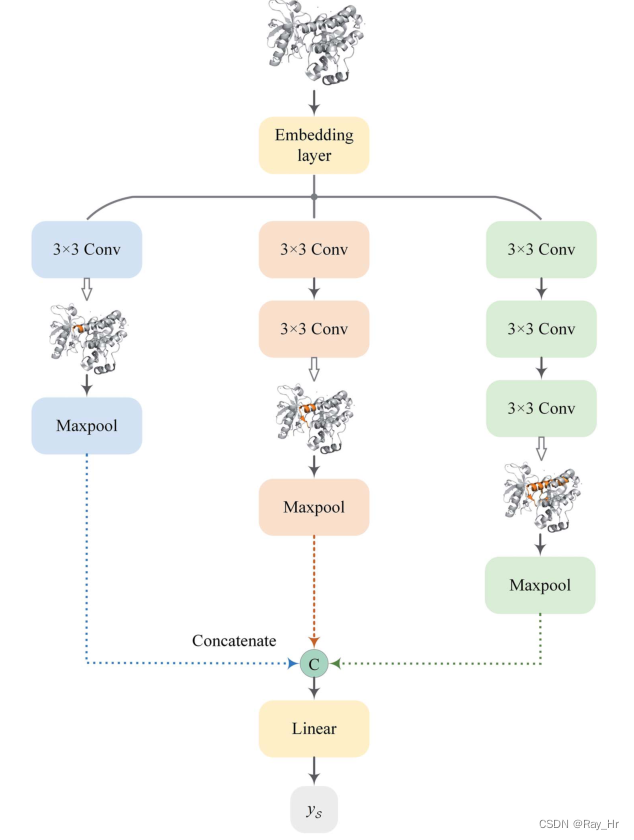
MCNN思路与MGNN类似,用不同感受野的卷积层来捕获不同尺度信息。通过堆叠3*3的卷积层来增大感受野,分别用了3、5、7层卷积来捕获蛋白质结构信息,并且只有部分蛋白质结构对DTA预测有作用,所以不需要像MGNN考虑捕获全局特性。对于输入长度为1200的蛋白质序列,用embedding层将其转换为128维向量,之后使得输入矩阵为S ∈ R 1200 × 128 \mathcal{S} \in \mathrm{R}^{1200 \times 128}S ∈R 1200 ×128(此处不太明白,转换后的矩阵是指特征变为128?),之后使用MCNN将输入矩阵转化为特征向量y S ∈ R d y_{\mathcal{S}} \in \mathbb{R}^{d}y S ∈R d。
y S = W ( m ( F 1 ( S ) ) ∥ m ( F 2 ( S ) ) ∥ m ( F 3 ( S ) ) ) y_{\mathcal{S}}=W\left(m\left(\mathcal{F}{1}(\mathcal{S})\right)\left\|m\left(\mathcal{F}{2}(\mathcal{S})\right)\right\| m\left(\mathcal{F}{3}(\mathcal{S})\right)\right)y S =W (m (F 1 (S ))∥m (F 2 (S ))∥m (F 3 (S )))
其中F i \mathcal{F}{i}F i 是一个3 * 3的卷积层,并且每个卷积层后加了激活函数ReLU,m表示最大值池化操作将C转化为 h 维向量,并且W ∈ R d × 3 h W \in \mathbb{R}^{d \times 3 h}W ∈R d ×3 h为可学习矩阵。
; 3.5 MGraphDTA network architecture
在上述的操作后,得到了可表示药物和靶点的向量,将两向量串联后输入多层感知机即可预测亲和力。MLP主要由三个线性变换层组成,每一层都通过激活函数和dropout层(0.1的dropout rate)。总得来讲,MGraphDTA包含一个MGNN用于药物编码,一个MCNN用于目标编码(蛋白质)。用均方误差来作为损失函数:
M S E = 1 n ∑ i = 1 n ( P i − Y i ) 2 \mathrm{MSE}=\frac{1}{n} \sum_{i=1}^{n}\left(P_{i}-Y_{i}\right)^{2}MSE =n 1 ∑i =1 n (P i −Y i )2
其中P i P_i P i 是药物靶点对应预测值,Y i Y_i Y i 是真实值,n是样本数。也可以简单的用交叉熵损失取替换MSE做二元分类问题(有效/无效)。
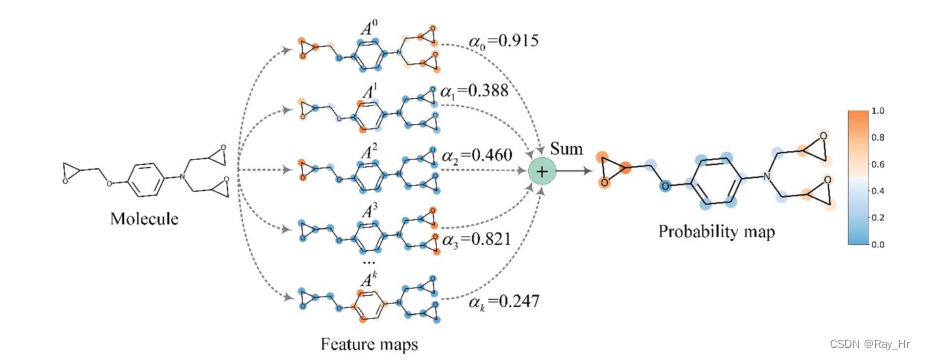
3.6 gradient-weighted affinity activation mapping
这部分就介绍了提升DTA模型的可解释性,用最后一层图卷积的输入梯度来表示每一个神经元的亲和力,由于图卷积层本身就可以保留全连接层中的空间信息,因此本文认为最后一个图卷积层在高级语义和详细的空间信息中有着最佳折中。将最后一个图卷积层的特征图表示为A,化学概率图P G r a d − A A M ∈ R V P_{G r a d-A A M} \in \mathbb{R}^{V}P G r a d −AA M ∈R V用给定分子的顶点数来表示。首先计算在第k个通道和第v个顶点处的神经元A v k A_{v}^{k}A v k 的亲和力得分的梯度,之后计算通道重要性权重α k \alpha_{k}αk
α k = 1 ∣ V ∣ ∑ v ∈ V ∂ P ∂ A v k \alpha_{k}=\frac{1}{|\mathcal{V}|} \sum_{v \in \mathcal{V}} \frac{\partial P}{\partial A_{v}{ }^{k}}αk =∣V ∣1 ∑v ∈V ∂A v k ∂P
之后进行加权聚合以及ReLU层后
P Grad-AAM = ∑ k α k A k P_{\text {Grad-AAM }}=\sum_{k} \alpha_{k} A^{k}P Grad-AAM =∑k αk A k
最后将其归一化为[0,1],该图可以认为是GNN捕获分子重要几何结构的加权聚集
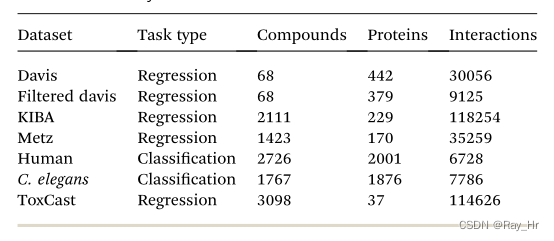
; 3.7 数据集(待补充)
4. 实验结果(待补充)
4.1 分类任务
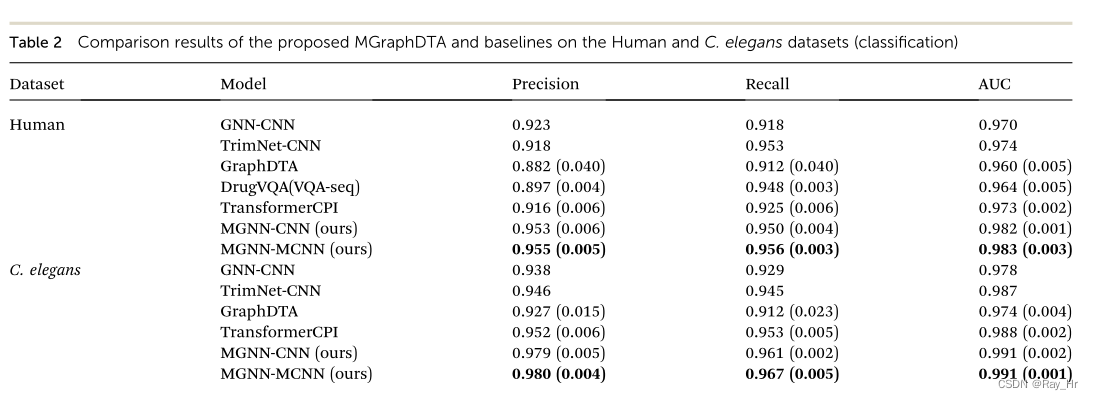
; 4.2 回归任务
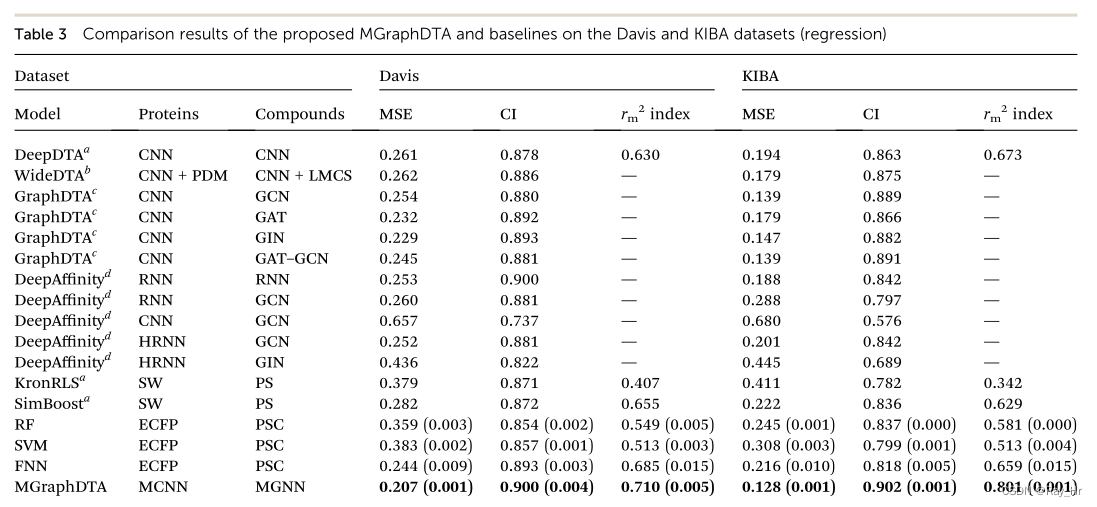
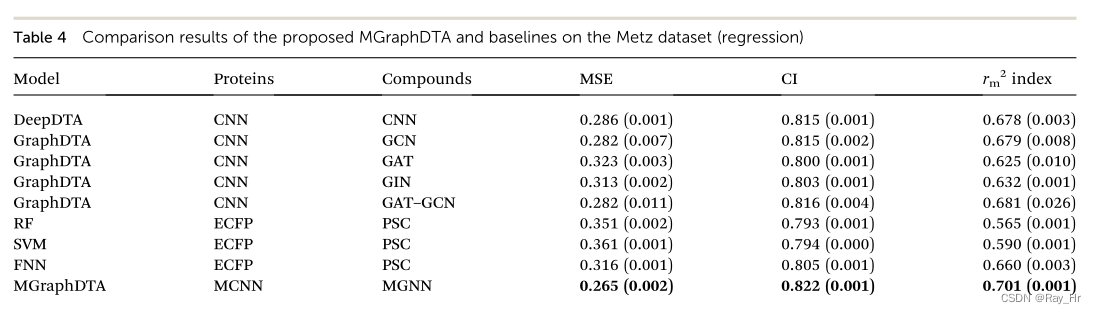
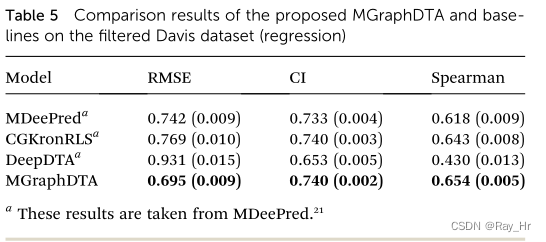
4.3 在更真实条件下的效果评估
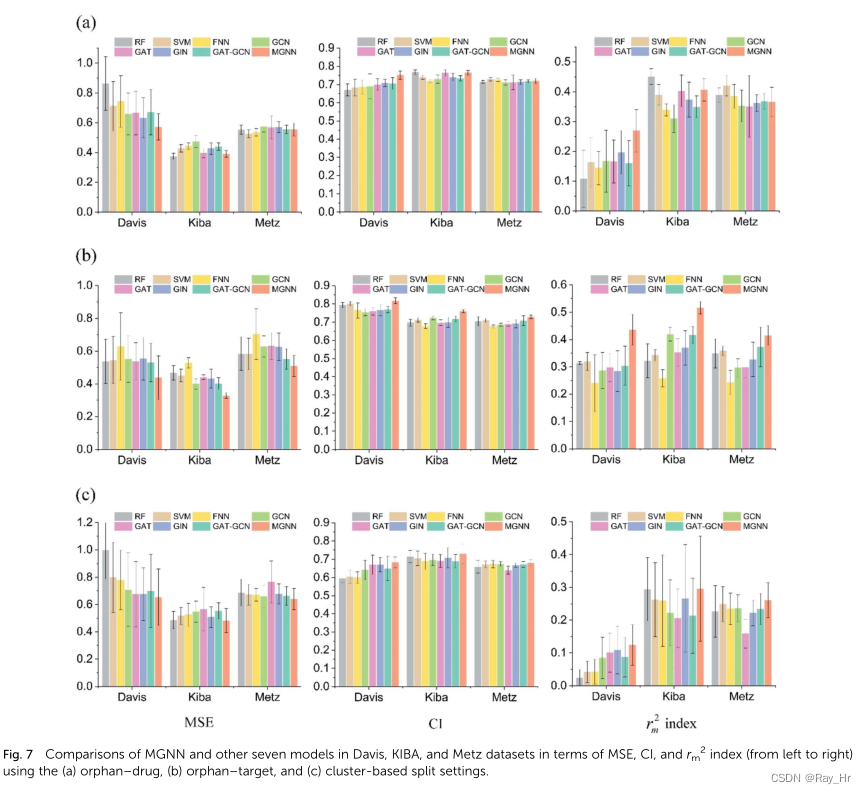
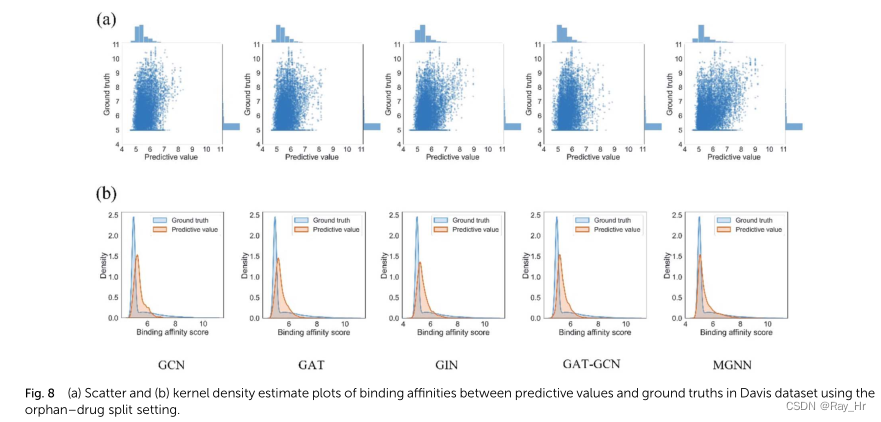
; 4.4 模型简化测试
4.5 视觉解释性
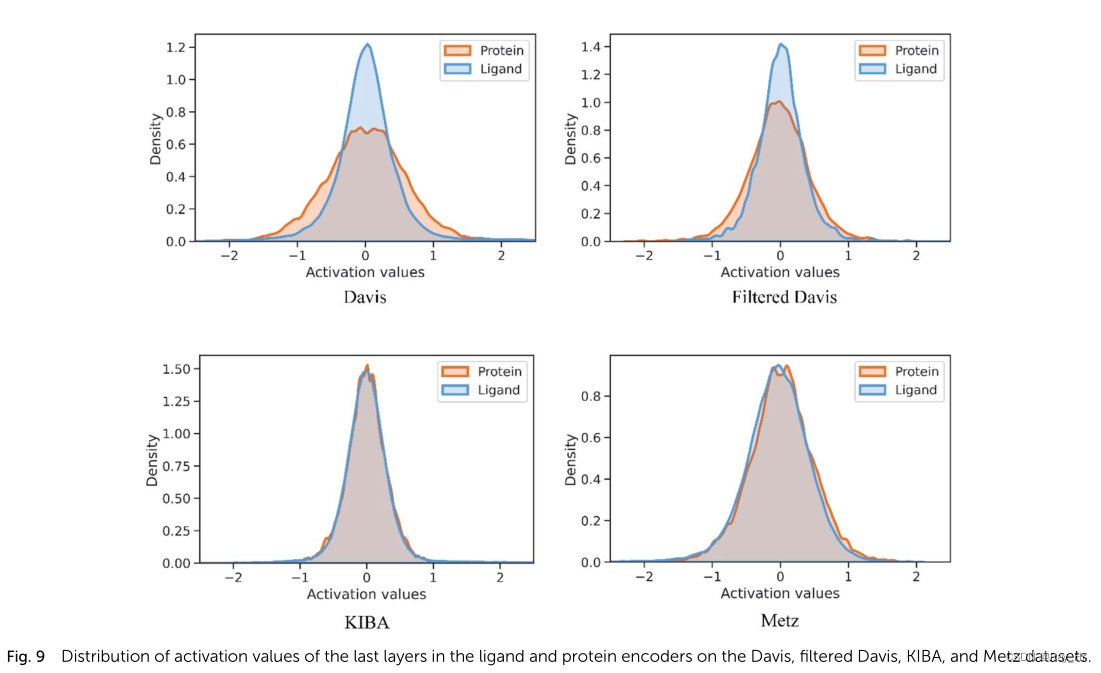

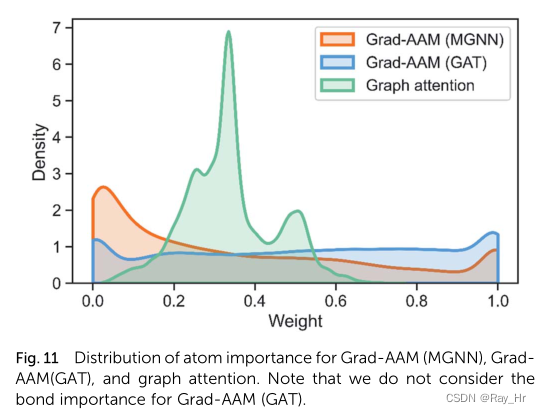
; 4.6 MGNN如何克服过平滑问题
深层的GNN将导致顶点的过平滑问题(过拟合?),而我们追求的模型需要深层网络以增大感受野,并且更具表现力。message pass机制是将从邻居收集的embedding 向量并与顶点特征组合完成更新,因此过度平滑表现为顶点embedding之间的相似性。随着模型层数的增加,顶点所表示的原子越来越多,因此两个不同顶点的embedding会变得越来越相似
4.7 局限性
5 结论
本文提出了一种基于化学直觉的新型图模型框架用于DTA预测:MGraphDTA.该模型用具有27层图卷积的MGNN来捕获分子多尺度结构,利用GradAAM进行可视化解释。
Original: https://blog.csdn.net/qq_39917739/article/details/126200548
Author: Ray_Hr
Title: 【无标题】
原创文章受到原创版权保护。转载请注明出处:https://www.johngo689.com/690170/
转载文章受原作者版权保护。转载请注明原作者出处!