

对一篇影像组学的的论文(《Development and validation of an MRI-based radiomics nomogram for distinguishing Warthin’s tumour from pleomorphic adenomas of the parotid gland》)中方法进行复现。完整地跑通影像组学全流程,对临床+影像组学特征进行建模并绘制Lasso回归图和ROC曲线、诺模图、校准曲线、决策曲线等。
需要完整代码到这里https://aistudio.baidu.com/aistudio/projectdetail/3270880 fork项目之后下载glioma.7z压缩包,然后用RStudio的RMarkdown进行运行
1.项目介绍
2012年荷兰学者提出影像组学(radiomics)概念:借助计算机,从医学影像中挖掘海量定量影像特征,使用统计学/机器学习方法,筛选出最有价值的影像学特征,用来解析临床信息。
可以说影像组学是人工智能在医学领域的一种特定研究方式。

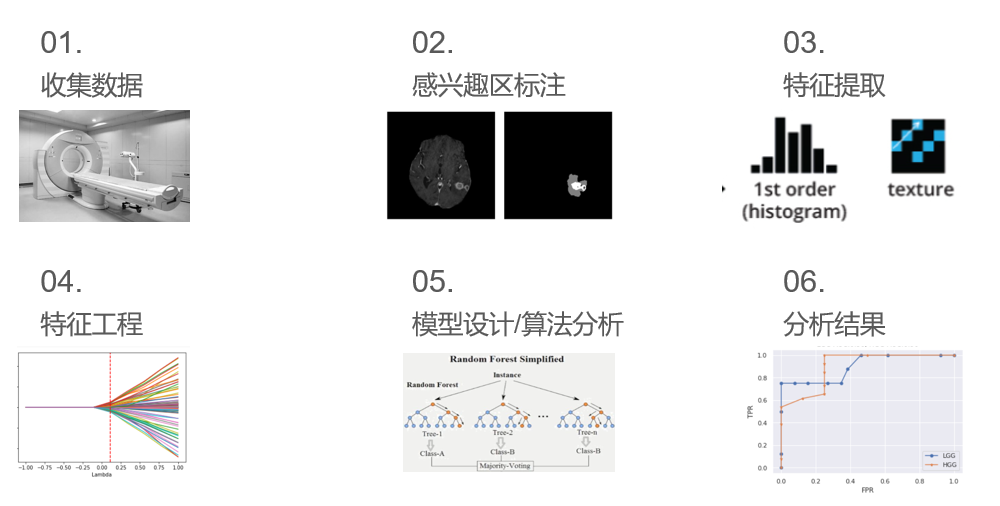
做影像组学一般会经过以下步骤1.收集数据,2.标注数据,3.特征提取,4.特征工程,5.模型设计,6.结果分析和绘图。
; 1.1 项目目的
而对于大多数医学专业的朋友来说,毕竟学医的对写代码不一定擅长,或者第一次接触影像组学。因此想打算写一个影像组学相关的项目,对某篇组学论文方法进行复现。通用这个项目的代码希望可以稍微帮助到他们,可以让他们把时间更多花在疾病的分析上,而不是解决某个bug花大量的时间。
1.2 项目大概情况
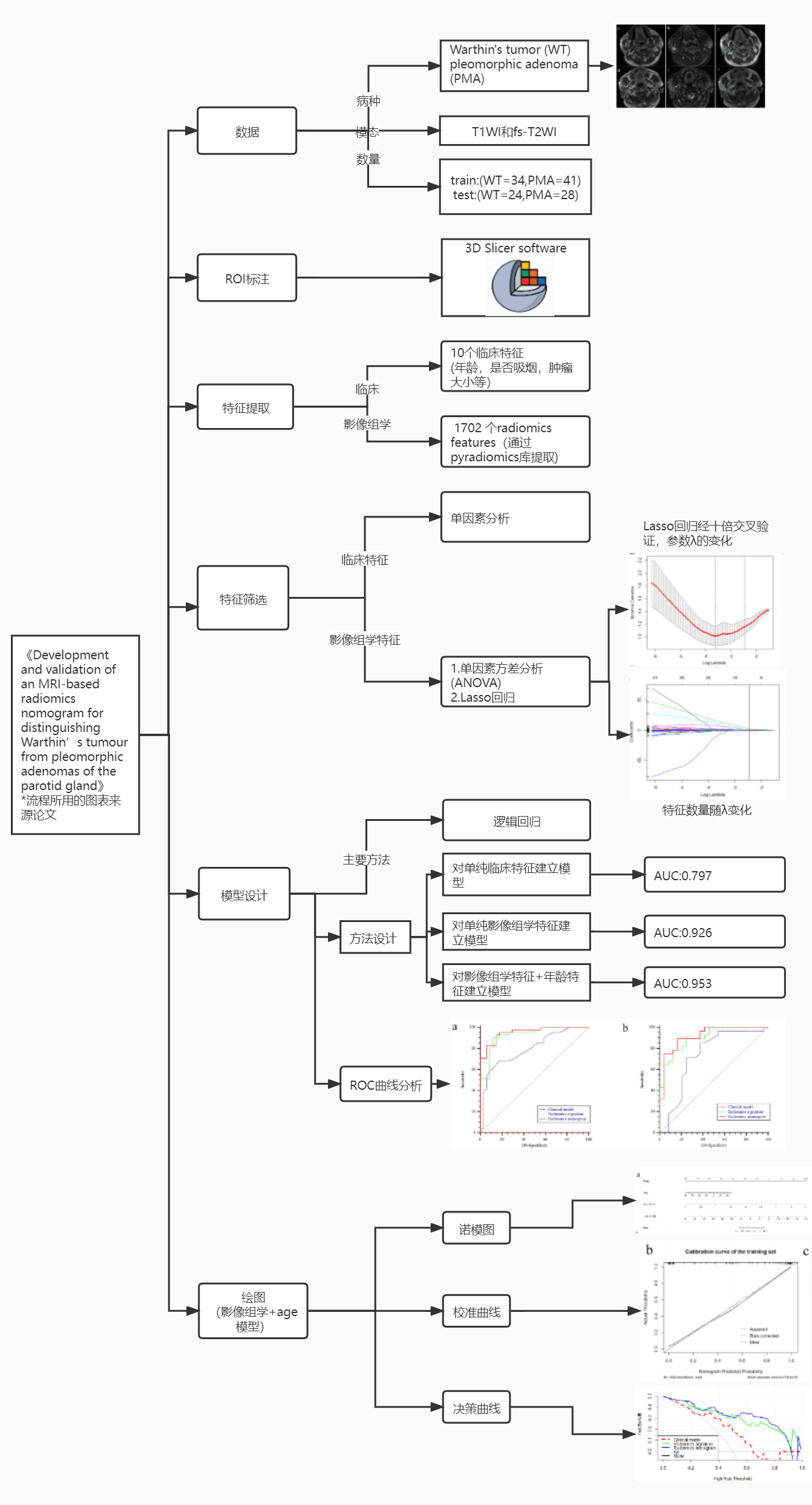
这个项目主要是对《Development and validation of an MRI-based radiomics nomogram for distinguishing Warthin’s tumour from pleomorphic adenomas of the parotid gland》这篇影像组学论文中的方法进行复现,完整的跑一个影像组学流程。包括:
1.对临床特征进行建模
2.提取影像组学特征,通过LassoCv进行特征筛选再建模
3.结合影像组学特征+age临床特征进行建模
4.绘制三个模型的ROC曲线进行对比
5.对组合模型绘制诺模图+决策曲线+校准曲线
1.3运行环境
由于有些图例,例如诺模图(列线图)需要用到R语言来绘制,但是提取特征用到python语言的Pyradiomics库,所以这个项目是一个混合编程语言的项目。刚好RMarkdown可以同时运行Python和R语言,所以请有需要运行项目的请离线安装RMarkdown来运行。
虽然不能通过在线BML运行项目,但是可以展示复现论文方法的代码,主要可以容易复制粘贴代码~~~ 。
注:对于如何安装python和安装R语言和如何使用RMarkdown不是这篇项目的范围,自己通过百度自学学习。
注:这个项目也是我在学习影像组学中做下的笔记,如有错误请纠正和谅解
2.关于数据
虽然是论文复现,但是论文中涉及的腮腺MR数据是不公开的,无法和论文实验数据一致,不过这个项目主要是为了实现论文中大部分方法。因此采用了一个公开分割比赛的数据集作为这个项目的数据。
在数据上用到的是一个公开的胶质瘤数据集–【BraTS2019】。是一个两分类的数据集,Hgg是高级别胶质瘤,Lgg是低级别的胶质瘤,数量上分别是240和76。每个病例有四个模态的磁共振图像。Hgg患者是带有age的年龄数据,但是lgg的患者是没有的。为了不影响临床+影像组学的建模。在(25到55)之间随机生成一个随机数赋予个Lgg患者作为他的年龄。所以Lgg患者的年龄是虚拟数据。提取影像组学的特征只用到T1增强的模态。
因为原始的数据集Hgg和Lgg的比例差不多达到3比1,所以为了搭到数据均衡一点,把Hgg删掉一部分,剩下101例。Lgg为76例。
因此在实际运用到自己的数据的时候,收集病例尽量做到数据均衡一点。不然有点自己坑自己的感觉。

; 3.复现的论文
《Development and validation of an MRI-based radiomics nomogram for distinguishing Warthin’s tumour from pleomorphic adenomas of the parotid gland》
3.1论文地址
3.2论文大概内容
这篇论文主要对腮腺肿瘤(Warthin和多形性腺瘤)的磁共振图像使用计算机提取大量高通量的特征,通过对特征进行筛选和建立模型来鉴别MR图像是Warthin还是多形性腺瘤。
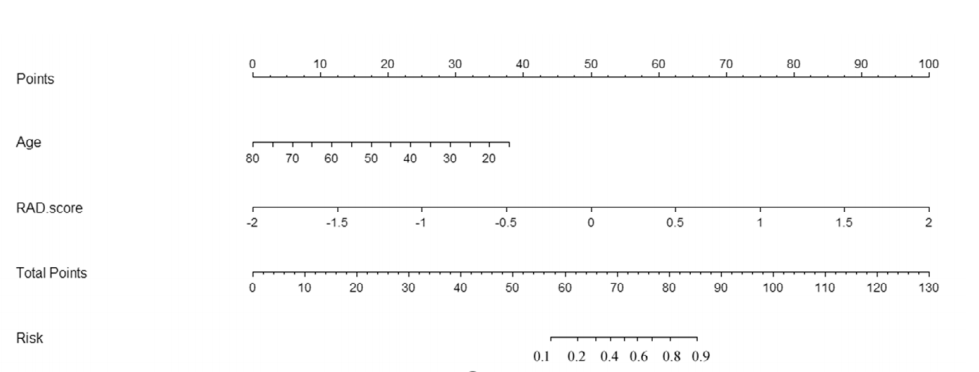
两种腺瘤的治疗方法和预后不同,建立一个良好的分类模型对临床来说至关重要。论文通过建立三种不同特征的模型,分别是1.单纯临床特征模型,2.单纯影像组学特征模型,3.影像组学特征+年龄模型。经过实验得到模型3的auc最好。并根据模型3构建影像组学列线图(如下图),使预测模型的结果更具有可读性,让模型更加方便运用实际临床决策之中。
; 3.2论文中的方法流程:
4.开始影像组学
4.1目录结构和数据准备
重要的一点就是临床特征的index列需要和每一例病例文件夹要一一对应
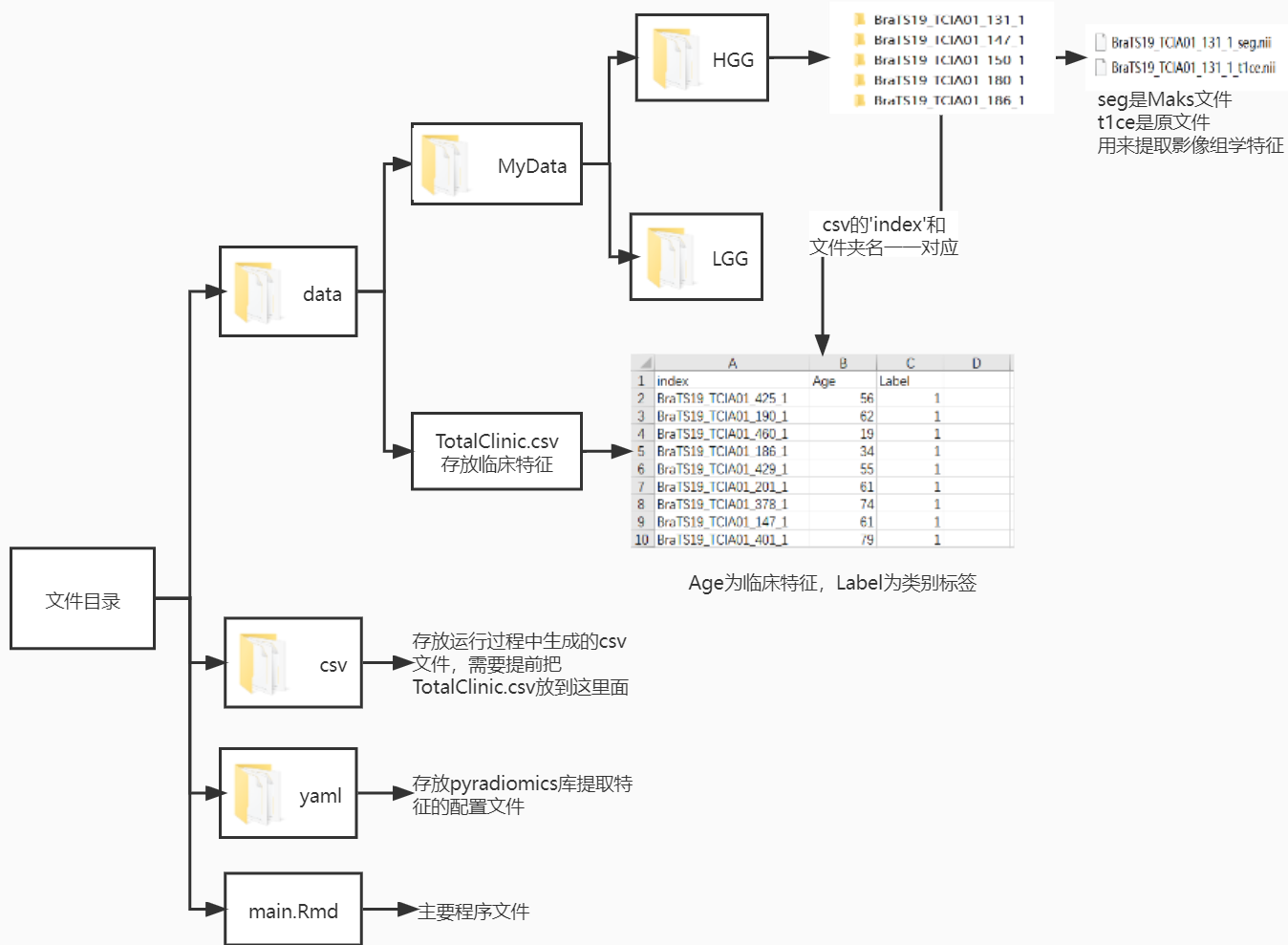
; 4.2 特征提取
特征提取是采用pyradiomics库进行提取,可以通过配置文件来设置提取那些特征,例如设置提取特征前是否重采样,设置滤波核的大小等等。如下图
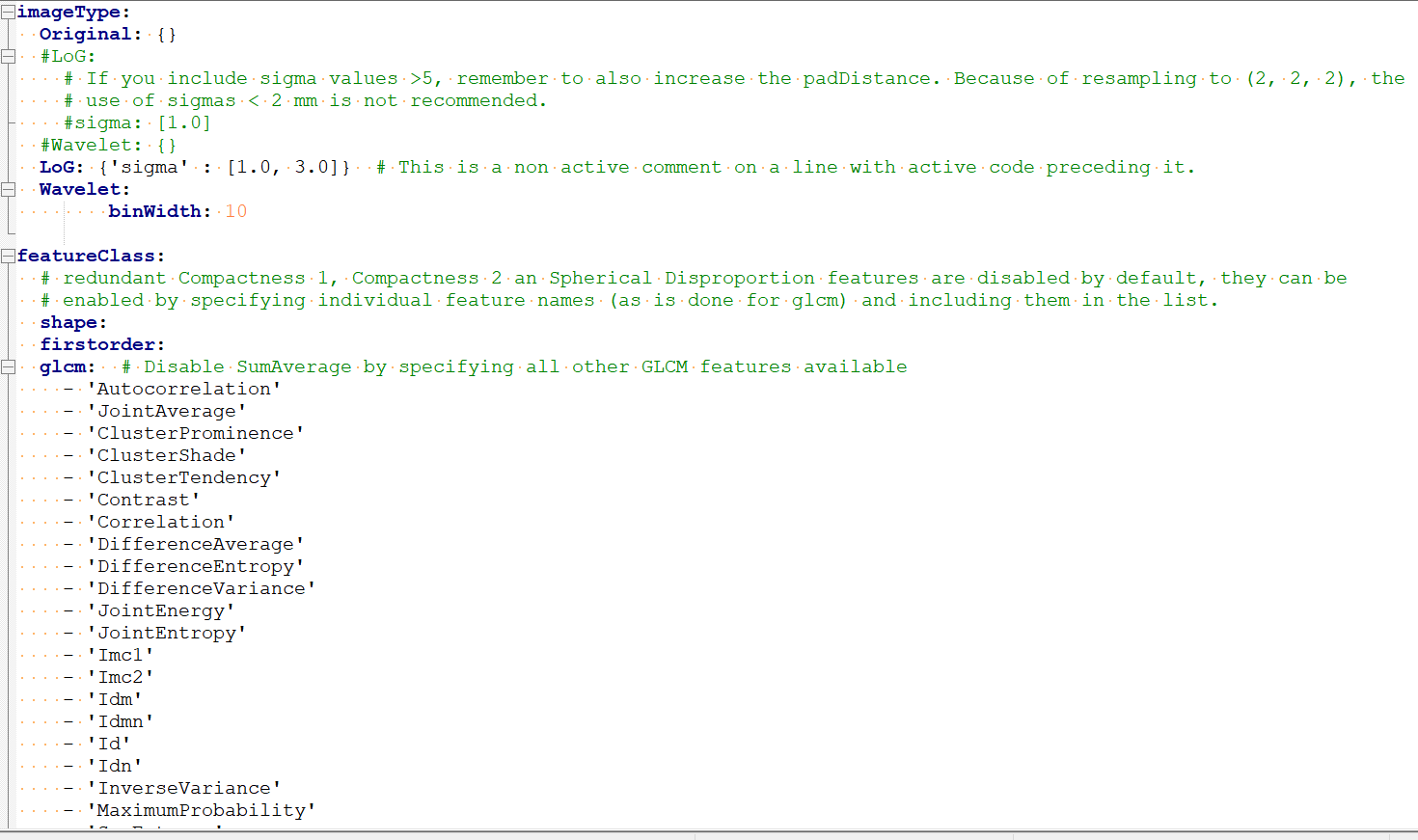
可以到pyradiomics官网查看地址
"""
经过一两个小时提取后会生成HGG.csv和LGG.csv文件,
生成的csv文件每一行都有接近一千个特征,数量会根据不同yaml文件设置不同而不同
"""
import sys
import pandas as pd
import os
import random
import shutil
import numpy as np
import radiomics
from radiomics import featureextractor
import SimpleITK as sitk
kinds = ['HGG','LGG']
para_path = 'yaml/MR_1mm.yaml'
extractor = featureextractor.RadiomicsFeatureExtractor(para_path)
dir = 'data/MyData/'
for kind in kinds:
print("{}:开始提取特征".format(kind))
features_dict = dict()
df = pd.DataFrame()
path = dir + kind
for index, folder in enumerate( os.listdir(path)):
for f in os.listdir(os.path.join(path, folder)):
if 't1ce' in f:
ori_path = os.path.join(path,folder, f)
break
lab_path = ori_path.replace('t1ce','seg')
features = extractor.execute(ori_path,lab_path)
features_dict['index'] = folder
for key, value in features.items():
features_dict[key] = value
df = df.append(pd.DataFrame.from_dict(features_dict.values()).T,ignore_index=True)
print(index)
df.columns = features_dict.keys()
df.to_csv('csv/' +'{}.csv'.format(kind),index=0)
print('Done')
print("完成")
"""
再对HGG.csv和LGG.csv文件进行处理,去掉字符串特征,插入label标签。
HGG标签为1,LGG标签为0
"""
import matplotlib.pyplot as plt
import seaborn as sns
hgg_data = pd.read_csv('csv/HGG.csv')
lgg_data = pd.read_csv('csv/LGG.csv')
hgg_data.insert(1,'label', 1)
lgg_data.insert(1,'label', 0)
cols=[x for i,x in enumerate(hgg_data.columns) if type(hgg_data.iat[1,i]) == str]
cols.remove('index')
hgg_data=hgg_data.drop(cols,axis=1)
cols=[x for i,x in enumerate(lgg_data.columns) if type(lgg_data.iat[1,i]) == str]
cols.remove('index')
lgg_data=lgg_data.drop(cols,axis=1)
total_data = pd.concat([hgg_data, lgg_data])
total_data.to_csv('csv/TotalOMICS.csv',index=False)
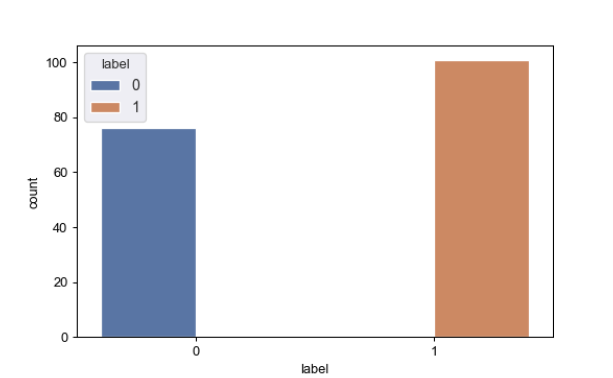
fig, ax = plt.subplots()
sns.set()
ax = sns.countplot(x='label',hue='label',data=total_data)
plt.show()
print(total_data['label'].value_counts())
4.3划分数据
对影像组学数据集进行划分训练集合测试集。比例为8:2。同样把临床特征也进行同样的划分(顺序和影响组学特征是一致的)。
library(glmnet)
library(rms)
library(foreign)
library(ggplot2)
library(pROC)
set.seed(888)
data <- read.csv("csv/TotalOMICS.csv")
nn=0.8
print(paste('总样本数:',length(data[,1])))
sub<-sample(1:nrow(data),round(nrow(data)*nn))
trainOmics<-data[sub,]
testOmics<-data[-sub,]
print(paste('训练集样本数:',length(trainOmics[,1])))
print(paste('测试集样本数:',length(testOmics[,1])))
write.csv(trainOmics,"csv/trainOmics.csv",row.names = FALSE )
write.csv(testOmics,"csv/testOmics.csv",row.names = FALSE )
"""
R语言输出结果
[1] "总样本数: 177"
[1] "训练集样本数: 142"
[1] "测试集样本数: 35"
"""
"""
根据上面对影像组学TotalOMICS.csv的数据划分,对TotalClinic.csv同样的顺序划分
"""
import pandas as pd
def compare(data1, data2,filename):
dt1 = pd.read_csv(data1,encoding='utf-8')
dt2 = pd.read_csv(data2,encoding='gb18030')
dt2.head()
df = pd.DataFrame()
dt1_name = dt1['index'].values.tolist()
dt2_name = dt2['index'].values.tolist()
for i in dt1_name:
if i in dt2_name:
dt2_row = dt2.loc[dt2['index'] == i]
df = df.append(dt2_row)
df.to_csv('./csv/'+filename+'.csv',header=True,index=False,encoding="utf_8_sig")
data_train= "./csv/trainOmics.csv"
data_test = "./csv/testOmics.csv"
data_clinic= "./csv/TotalClinic.csv"
compare(data_train,data_clinic,"trainClinic")
compare(data_test,data_clinic,"testClinic")
4.4对单纯临床特征进行建模
因为这个数据集只有age这个临床特征,所以没有做进一步的特征分析和筛选,直接用age进行建模
4.4.1处理临床数据
trainClinic<-read.csv("./csv/trainClinic.csv",fileEncoding = "UTF-8-BOM")
trainClinic<- data.frame(trainClinic)
trainClinic$Age <- as.numeric(trainClinic$Age)
names(trainClinic)
"""
R语言输出结果
[1] "index" "Age" "Label"
"""
4.4.2 使用逻辑回归对临床特征进行建模
model.Clinic<-glm(Label~Age,data = trainClinic,family=binomial(link='logit'))
summary(model.Clinic)
"""
R语言输出结果
Call:
glm(formula = Label ~ Age, family = binomial(link = "logit"),
data = trainClinic)
Deviance Residuals:
Min 1Q Median 3Q Max
-1.7106 -0.6673 0.1870 0.6294 2.6505
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -6.6037 1.1252 -5.869 4.39e-09 ***
Age 0.1419 0.0233 6.090 1.13e-09 ***
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 194.566 on 141 degrees of freedom
Residual deviance: 84.983 on 136 degrees of freedom
AIC: 96.983
Number of Fisher Scoring iterations: 6
"""
4.6.1 查看在训练集的情况
probOmicsTrain<-predict.glm(object =model.Omics,newdata=tData_train,type = "response")
predOmicsTrain<-ifelse(probOmicsTrain>=0.5,1,0)
error=predOmicsTrain-tData_train$label
accuracy=(nrow(tData_train)-sum(abs(error)))/nrow(tData_train)
precision=sum(tData_train$label & predOmicsTrain)/sum(predOmicsTrain)
recall=sum(predOmicsTrain & tData_train$label)/sum(tData_train$label)
F_measure=2*precision*recall/(precision+recall)
print(paste('准确率accuracy:',accuracy))
print(paste('精确率precision:',precision))
print(paste('召回率recall:',recall))
print(paste('F_measure:',F_measure))
table(tData_train$label,predOmicsTrain)
"""
R语言输出结果
[1] "准确率accuracy: 0.908450704225352"
[1] "精确率precision: 0.935064935064935"
[1] "召回率recall: 0.9"
[1] "F_measure: 0.917197452229299"
predOmicsTrain
0 1
0 57 5
1 8 72
"""
4.6.2 查看模型在测试集的结果
tData_test <- read.csv("csv/testOmics.csv",fileEncoding = "UTF-8-BOM")
probOmicsTest<-predict.glm(object =model.Omics,newdata=tData_test,type = "response")
predOmicsTest<-ifelse(probOmicsTest>=0.5,1,0)
error=predOmicsTest-tData_test$label
accuracy=(nrow(tData_test)-sum(abs(error)))/nrow(tData_test)
precision=sum(tData_test$label & predOmicsTest)/sum(predOmicsTest)
recall=sum(predOmicsTest & tData_test$label)/sum(tData_test$label)
F_measure=2*precision*recall/(precision+recall)
print(paste('准确率accuracy:',accuracy))
print(paste('精确率precision:',precision))
print(paste('召回率recall:',recall))
print(paste('F_measure:',F_measure))
table(tData_test$label,predOmicsTest)
"""
R语言输出结果
[1] "准确率accuracy: 0.857142857142857"
[1] "精确率precision: 0.863636363636364"
[1] "召回率recall: 0.904761904761905"
[1] "F_measure: 0.883720930232558"
predOmicsTest
0 1
0 11 3
1 2 19
"""
4.7结合临床特征和影像组学特征进行建模
先处理数据,把临床特征的age赋值到影像组学中新增age一列。然后根据lasso模型计算每个病例的得分。把这个得分和age作为新的特征建立逻辑回归模型。
4.7.1处理数据
tData_train2 <- read.csv("csv/tData_train.csv",fileEncoding = "UTF-8-BOM")
tData_train2$Age <- trainClinic$Age
for (i in trainClinic$index){
if(tData_train2[tData_train2$index == i,]$index == i){
Age <- trainClinic[trainClinic$index == i,]$Age
tData_train2[tData_train2$index == i,]$Age <- Age
}
}
tData_test2 <- read.csv("csv/testOmics.csv",fileEncoding = "UTF-8-BOM")
testClinic2<-read.csv("./csv/testClinic.csv",fileEncoding = "UTF-8-BOM")
testClinic2<- data.frame(testClinic2)
testClinic2$Age <- as.numeric(testClinic2$Age)
columns <- colnames(tData_train2)[1:564]
tData_test2 <- as.data.frame(tData_test2[,columns])
tData_test2$Age <- testClinic2$Age
for (i in testClinic2$index){
if(tData_test2[tData_test2$index == i,]$index == i){
Age <- testClinic2[testClinic2$index == i,]$Age
tData_test2[tData_test2$index == i,]$Age <- Age
}
}
4.7.2根据lasso模型计算得分
y_vad <-as.data.frame(tData_test2$label)
y_vad <- model.matrix(~.,data=y_vad)[,-1]
yavars<-names(tData_test2) %in% c("label")
x_vad <- as.data.frame(tData_test2[!yavars])
columns <- colnames(tData_train2)[3:564]
x_vad <- as.data.frame(tData_test2[,columns])
x_vad <- model.matrix(~.,data=x_vad)[,-1]
x_dev <- X
y_dev <- Y
tData_train2$RS <-predict(fit,type="link",
newx=x_dev,newy=y_dev,s=cv.fit$lambda.1se)
tData_test2$RS<-predict(fit,type="link",
newx=x_vad,newy=y_vad,cv.fit$lambda.1se)
4.7.3建立逻辑回归模型
tData_train2$RS <- as.numeric(tData_train2$RS)
model.and1 = glm(label ~ RS+Age,data=tData_train2,binomial(link='logit'))
print(model.and1$coef)
print("#####")
model.and2 <- lrm(label ~ RS+Age,data=tData_train2,x=TRUE,y=TRUE)
print(model.and2$coef)
"""
R语言输出结果
(Intercept) RS Age
-5.8730358 2.4543423 0.1192463
[1] "#####"
Intercept RS Age
-5.8730331 2.4543415 0.1192462
"""
4.7.4查看模型在临床结合影像组学的训练集的情况
probOmicsTrainAnd<-predict.glm(object =model.and1,newdata=tData_train2,type = "response")
predOmicsTrainAnd<-ifelse(probOmicsTrainAnd>=0.5,1,0)
error=predOmicsTrainAnd-tData_train2$label
accuracy=(nrow(tData_train2)-sum(abs(error)))/nrow(tData_train2)
precision=sum(tData_train2$label & predOmicsTrainAnd)/sum(predOmicsTrainAnd)
recall=sum(predOmicsTrainAnd & tData_train2$label)/sum(tData_train2$label)
F_measure=2*precision*recall/(precision+recall)
print(paste('准确率accuracy:',accuracy))
print(paste('精确率precision:',precision))
print(paste('召回率recall:',recall))
print(paste('F_measure:',F_measure))
table(tData_train2$label,predOmicsTrainAnd)
"""
R语言输出结果
[1] "准确率accuracy: 0.929577464788732"
[1] "精确率precision: 0.948717948717949"
[1] "召回率recall: 0.925"
[1] "F_measure: 0.936708860759494"
predOmicsTrainAnd
0 1
0 58 4
1 6 74
"""
4.7.5 查看模型在临床结合影像组学的测试集的情况
tData_test2$RS <- as.numeric(tData_test2$RS)
probOmicsTestAnd<-predict.glm(object =model.and1,newdata=tData_test2,type = "response")
predOmicsTestAnd<-ifelse(probOmicsTestAnd>=0.5,1,0)
error=predOmicsTestAnd-tData_test2$label
accuracy=(nrow(tData_test2)-sum(abs(error)))/nrow(tData_test2)
precision=sum(tData_test2$label & predOmicsTestAnd)/sum(predOmicsTestAnd)
recall=sum(predOmicsTestAnd & tData_test2$label)/sum(tData_test2$label)
F_measure=2*precision*recall/(precision+recall)
print(paste('准确率accuracy:',accuracy))
print(paste('精确率precision:',precision))
print(paste('召回率recall:',recall))
print(paste('F_measure:',F_measure))
table(tData_test2$label,predOmicsTestAnd)
"""
R语言输出结果
[1] "准确率accuracy: 0.857142857142857"
[1] "精确率precision: 0.9"
[1] "召回率recall: 0.857142857142857"
[1] "F_measure: 0.878048780487805"
predOmicsTestAnd
0 1
0 12 2
1 3 18
"""
4.8绘制ROC
把三个模型都绘制到同一个ROC曲线里面,进行比较
4.8.1绘制训练集的ROC曲线
rocClinic <- roc(trainClinic$Label, probClinicTrain)
rocOmics <- roc(tData_train$label, probOmicsTrain)
rocClinicAndOmics <-roc(tData_train2$label, probOmicsTrainAnd)
plot(rocClinic,
print.auc=TRUE,
print.auc.x=0.4, print.auc.y=0.6,
auc.polygon=TRUE,
auc.polygon.col="#fff7f7",
grid=FALSE,
print.thres=FALSE,
main=" Train ROC curves",
col="red",
legacy.axes=TRUE,
xlim=c(1,0),
mgp=c(1.5, 1, 0),
lty=3)
plot.roc(rocOmics,
add=TRUE,
col="green",
print.thres=FALSE,
print.auc=TRUE,
print.auc.x=0.4,print.auc.y=0.5,
lty=2)
plot.roc(rocClinicAndOmics,
add=TRUE,
col="blue",
print.thres=FALSE,
print.auc=TRUE,
print.auc.x=0.4,print.auc.y=0.4,
lty=1)
legend(0.45, 0.30,
bty = "n",
legend=c("Clinical model","Radiomics signature","Clinical and Radiomics"),
col=c("red","green","blue"),
lwd=1,
lty=c(3,2,1))

4.8.1绘制测试集的ROC曲线
rocClinicTest <- roc(testClinic$Label, probClinicTest)
rocOmicsTest <- roc(tData_test$label, probOmicsTest)
rocClinicAndOmicsTest <-roc(tData_test2$label, probOmicsTestAnd)
plot(rocClinicTest,
print.auc=TRUE,
print.auc.x=0.4, print.auc.y=0.6,
auc.polygon=TRUE,
auc.polygon.col="#fff7f7",
grid=FALSE,
print.thres=FALSE,
main=" Test ROC curves",
col="red",
legacy.axes=TRUE,
xlim=c(1,0),
mgp=c(1.5, 1, 0),
lty=3)
plot.roc(rocOmicsTest,
add=TRUE,
col="green",
print.thres=FALSE,
print.auc=TRUE,
print.auc.x=0.4,print.auc.y=0.5,
lty=2)
plot.roc(rocClinicAndOmicsTest,
add=TRUE,
col="blue",
print.thres=FALSE,
print.auc=TRUE,
print.auc.x=0.4,print.auc.y=0.4,
lty=1)
legend(0.45, 0.30,
bty = "n",
legend=c("Clinical model","Radiomics signature","Clinical and Radiomics"),
col=c("red","green","blue"),
lwd=1,
lty=c(3,2,1))
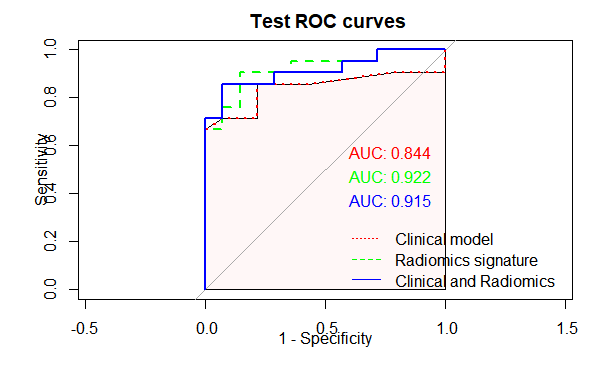
4.8绘制诺模图
临床结合影像组学建立的逻辑回归模型整体来说比较良好。
把这个模型绘制成诺模图,使模型可视化。
library(rms)
formula <- as.formula(label ~ Age+RS)
dd = datadist(tData_train2)
options(datadist="dd")
fitnomogram <- lrm(formula,data=tData_train2, x=TRUE, y=TRUE)
nom1 <- nomogram(fitnomogram,
fun=function(x)1/(1+exp(-x)),
lp=F,
fun.at=c(0.1,0.2,0.5,0.7,0.9),
funlabel="Risk")
plot(nom1)
诺模图怎样看?现在知道某个患者的age师多少岁,找到对应顶部Points的分数,同样知道患者的RS得分,也找到对应Points的得分,把两个等分加起来。在Total Points找到底部Risk得分,这个得分就是判断患者是HGG的概率。
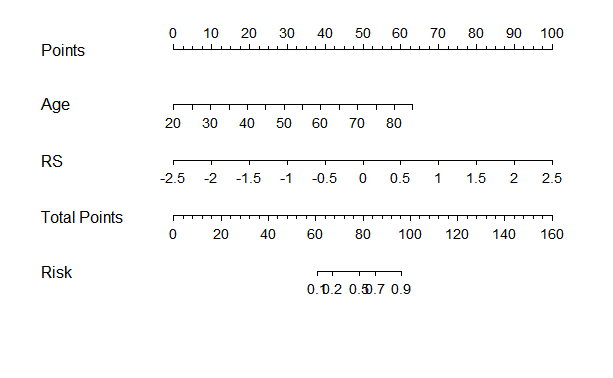
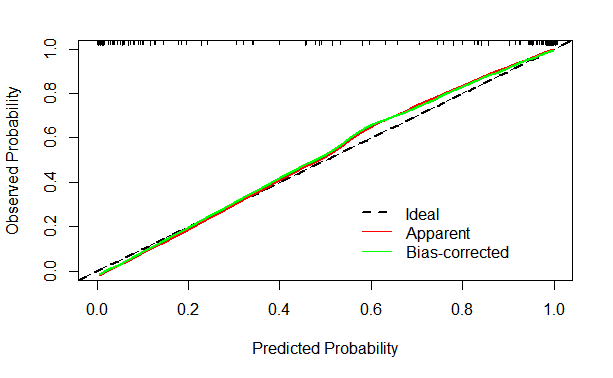
4.9绘制校准曲线
cal1 <- calibrate(fitnomogram, cmethod="hare", method="boot", B=1000)
plot(cal1, xlim=c(0,1.0), ylim=c(0,1.0),
xlab = "Predicted Probability",
ylab = "Observed Probability",
legend = FALSE,subtitles = FALSE)
abline(0,1,col = "black",lty = 2,lwd = 2)
lines(cal1[,c("predy","calibrated.orig")],
type = "l",lwd = 2,col="red",pch =16)
lines(cal1[,c("predy","calibrated.corrected")],
type = "l",lwd = 2,col="green",pch =16)
legend(0.55,0.35,
c("Ideal","Apparent","Bias-corrected"),
lty = c(2,1,1),
lwd = c(2,1,1),
col = c("black","red","green"),
bty = "n")
彩色的曲线越接近点斜线,模型的性能就比较好
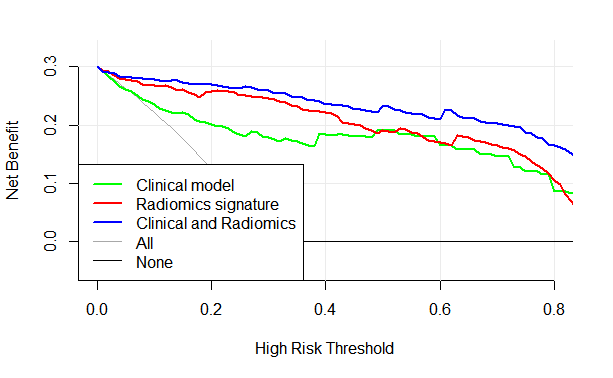
4.10 绘制决策曲线
把三个模型都绘制在同一个图中,进行比较
library(rmda)
formulClinic<-as.formula(Label~Age)
formulaOmics <- as.formula(label ~wavelet.LHL_glcm_Imc2+wavelet.LHL_glszm_ZoneEntropy+wavelet.HLL_glszm_ZoneEntropy+wavelet.LLL_firstorder_90Percentile+wavelet.LLL_firstorder_RootMeanSquared)
formulaAnd <- as.formula(label~Age+RS)
model_Clinic <- decision_curve(formulClinic, data=trainClinic,
family=binomial(link='logit'),
thresholds=seq(0,1,by=0.01),
confidence.intervals=0.95,
study.design = 'case-control',
population.prevalence=0.3)
model_Omics <- decision_curve(formulaOmics, data=tData_train,
family=binomial(link='logit'),
thresholds=seq(0,1,by=0.01),
confidence.intervals=0.95,
study.design = 'case-control',
population.prevalence=0.3)
model_And <- decision_curve(formulaAnd, data=tData_train2,
family=binomial(link='logit'),
thresholds=seq(0,1,by=0.01),
confidence.intervals=0.95,
study.design = 'case-control',
population.prevalence=0.3)
model_all <- list(model_Clinic,model_Omics,model_And)
plot_decision_curve(model_all,curve.names=c('Clinical model','Radiomics signature','Clinical and Radiomics'),
xlim=c(0,0.8),
cost.benefit.axis=F,col=c('green','red','blue'),
confidence.intervals = F,
standardize = F,
ain2,
family=binomial(link='logit'),
thresholds=seq(0,1,by=0.01),
confidence.intervals=0.95,
study.design = 'case-control',
population.prevalence=0.3)
model_all <- list(model_Clinic,model_Omics,model_And)
plot_decision_curve(model_all,curve.names=c('Clinical model','Radiomics signature','Clinical and Radiomics'),
xlim=c(0,0.8),
cost.benefit.axis=F,col=c('green','red','blue'),
confidence.intervals = F,
standardize = F,
legend.position="bottomleft")
一般曲线越靠左上角的,模型的性能就比较好,可以看到蓝色曲线(临床结合影像组学)的模型是在最外面的,可见这个模型性能比较好。
4.11 总结
到这一步基本结束这个项目,基本的方法和图表都已经绘制出来,代码都已经全部给出了。如果想在本地运行整个项目可以到这个项目地址(https://aistudio.baidu.com/aistudio/projectdetail/3270880)中下载【glioma.7z】压缩包,解压后用Rstudio软件打开就可以运行(前提已经配置好环境)
Original: https://blog.csdn.net/weixin_42990106/article/details/122102397
Author: 吖查
Title: 实现影像组学全流程
原创文章受到原创版权保护。转载请注明出处:https://www.johngo689.com/647965/
转载文章受原作者版权保护。转载请注明原作者出处!