

文章目录
- 论文概况
- 1. 研究背景
- 2. 研究数据
* - 2.1 种内数据集
- 2.2 种间数据集
- 2.3 多类别数据集
- 3. 研究方法
* - 3.1数据预处理
- 3.2局部特征提取
* - 3.3 结构特征提取
– - 3.4 预测模块
- 4. 结果
* - 4.1 种内数据集上的性能比较
– - 4.2 多物种数据集的性能比较
– - 4.3 多类别数据集性能比较
- 4.4 消融实验
– - 4.5 超参数的分析
– - 4.5.2 分类维度的选择
- 5. 结论
- 6. 创新点
- 7. 启发
论文概况
作者单位:湖南大学曾湘详课题组
发表期刊:《Briefings in Bioinformatics》,2020年期刊影响因子:11.622
发表时间:2022年1月11日
数据和代码:https://github.com/xzenglab/TAGPPI
- 研究背景
生物细胞的生命是由代谢和信号通路中的蛋白质相互作用控制的,通过对PPIs的深入了解,我们可以更深入地了解正常状态和疾病状态下的细胞生理学,促进相关任务,如靶向药物开发和治疗设计。蛋白质的空间结构与蛋白质的功能密切相关。整合蛋白质结构提高了蛋白质相互作用(PPI)预测的性能。然而,已知蛋白质结构的数量有限,限制了基于结构的预测方法的应用,利用预测的蛋白质结构信息是一种很有前途的方法来提高基于序列的预测方法的性能。我们提出了一种新的端到端框架TAGPPI仅使用蛋白质序列来预测PPIs。
- 研究数据
2.1 种内数据集
种内PPI数据集由酵母(Yeast)、大肠杆菌(E. coli)、秀丽隐杆线虫(C. elegans)和黑腹线虫(D.melanogaster)这四个数据集组成。酵母的种内PPI数据集是目前最先进的方法中广泛使用的基准数据集,所以本文大量的实现在酵母数据集上展开,如下表所示。
数据集蛋白质数量正样本数量负样本数量酵母(Yeast)249755945594大肠杆菌(E. coli)183469546954秀丽隐杆线虫(C. elegans)263740134013黑腹线虫(D.melanogaster)70582197521975
2.2 种间数据集
多物种数据集结合了种内的基准数据集。多物种数据集是用来评估模型预测不同物种间蛋白质相互作用的能力,这些蛋白质具有非常低的序列一致性,在这个数据集上训练和测试我们的模型,以估计TAGPPI在多种物种上的能力,蛋白质少于50个氨基酸或高序列鉴定(40%, 25%, 10%或1%)被移除,如下表所示。


; 2.3 多类别数据集
该数据集是由AlphaFold预测的Homo sapiens蛋白质结构的数据集,它们的标签是通过与STRING数据库进行匹配获得,蛋白质数量和样本数量分别为:16278和75875。数据集中有七种类型的相互作用:激活(activation,)、绑定(,binding)、催化(catalysis)、表达(expression)、抑制(inhibihtion)、翻译后修饰(post-translational modification)和反应(reaction)。
- 研究方法
本文提出了一个端到端的深度学习框架TAGPPI来识别PPI, PPI预测任务是一个基于多维蛋白质数据的二元分类问题。TAGPPI旨在学习一个映射函数f(.),该函数输入两个蛋白质的序列特征及其预测的接触图。然后来预测是否存在相互作用。该方法主要包括以下四步:
- 数据处理:用一种预先训练好的嵌入模型来初始化氨基酸
- 序列局部特征提取: 用序列特征学习模块在一维空间上捕获局部特征。
- 基于图的空间特征聚合:利用堆叠图神经网络和池化层选择空间结构特征。在接触图上实现了图形表示学习方法,获得了蛋白质的三维结构特征。
- 多层感知器的预测模块:将序列局部特征和图的空间特征进行结合,通过堆叠的三层全连接层完成最终的预测。
模型如下图所示
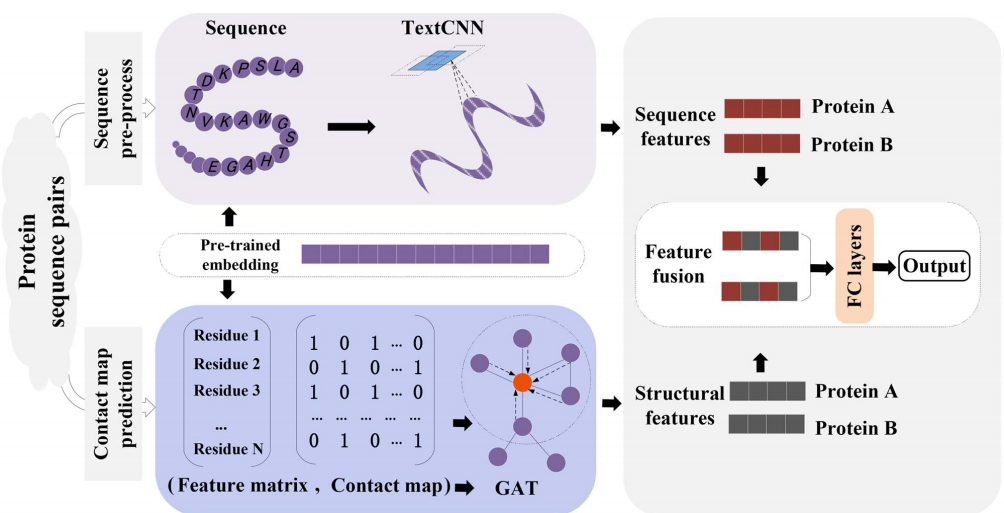
; 3.1数据预处理
利用氨基酸嵌入将蛋白质嵌入到不同的维度空间中,提取蛋白质的多维信息。利用SeqVec来实现氨基酸嵌入,SeqVec是通过训练ELMo语言模型得到的取自蛋白质序列Uniref50数据集,SeqVec采用字符序列嵌入和图节点嵌入两种方法实现了序列嵌入,嵌入的输出为X ∈ R L ∗ M X∈R^{L*M}X ∈R L ∗M。
3.2局部特征提取
通过3.1部分的SeqVec得到矩阵的维度为X ∈ R L ∗ M X∈R^{LM}X ∈R L ∗M, M表示选择的特征维度作者设置为1024,L表示蛋白质序列氨基酸的数量。为了确保TextCNN模块的输出矢量大小是固定的,取蛋白质序列的最大氨基酸数L=1200,长度小于1200则用0进行填充,得到的嵌入矩阵的大小为R 1200 ∗ 1024 R^{12001024}R 1 2 0 0 ∗1 0 2 4,使用一维卷积的TextCNN来捕捉蛋白质序列的局部特征,该TextCNN包括三个卷积层和三个 max-pooling,具体结构如下图所示:
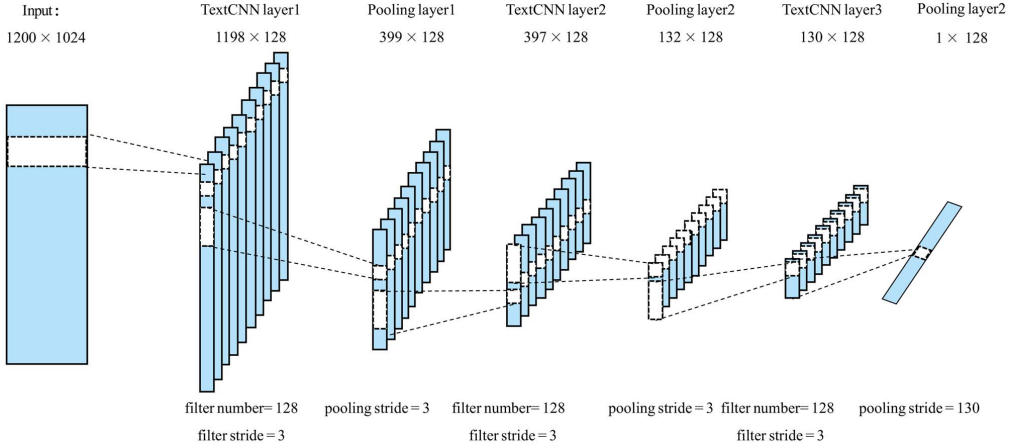
模型的参数如下:
- 三个卷积层参数:filter=128,kernel_size = 3,stide=1
- 前两个max-pooling:pool_size = 3,stide=3
- 最后一个max-pooling:pool_size =130,stide=1
; 3.3 结构特征提取
3.3.1 构建预测接触图
长度为L的蛋白质的接触图是一个方阵,当两个残基的C β C_ βC β原子的欧式距离小于8Å时判定两个残基是接触的,方阵定义如下公式:

AlphaFold能够以原子精度预测蛋白质结构,是DeepMind和EMBL的欧洲生物信息学研究所提供蛋白质结构预测,它建立在多年以前使用大量基因组数据(约 170,000 个蛋白质结构)来预测蛋白质结构的研究基础之上。 AlphaFold产生的蛋白质3D模型比以往任何一种都精确得多,在生物学的核心挑战之一上取得了重大进展。作者使用上述定义的规则在AlphaFold数据库中构建了接触图,并建立了几个用于PPI预测的接触图数据库。
; 3.3.2 图表示学习
蛋白质空间图定义为G = (V, A),其中V表示节点集,A ∈ R L ∗ L A∈R^{LL}A ∈R L ∗L表示邻接矩阵,通过 AlphaFold预测的接触图学习得到,为了学习蛋白质的结构特征,我们按照全maxpooling操作设计了三个堆叠的图注意网络(GATs)模块,如下图所示,GATs对邻居节点进行聚合时采用注意机制。对目标节点影响较大的邻居节点在聚合中会获得较大的权重系数,邻近氨基酸在空间上的依赖性,即使它们的顺序很远。图注意层的初始输入是一组节点特征h ( 0 ) = ( h 1 0 , h 2 0 , h 3 0 , h L 0 , h m 0 ∈ R d 0 ) h^{(0)}=({h_1^{0},h_2^{0},h_3^{0},h_L^{0},h_m^{0}∈R^{d^0})}h (0 )=(h 1 0 ,h 2 0 ,h 3 0 ,h L 0 ,h m 0 ∈R d 0 ),d 0 = 1024 {d^0}=1024 d 0 =1 0 2 4,利用氨基酸嵌入方法,在氨基酸嵌入中描述的一套新的节点特征,第l个GAT的节点特征表示为h ( l ) = ( h 1 l , h 2 l , h 3 l , h L l , h m l ∈ R d l ) h^{(l)}=({h_1^{l},h_2^{l},h_3^{l},h_L^{l},h_m^{l}∈R^{d^l})}h (l )=(h 1 l ,h 2 l ,h 3 l ,h L l ,h m l ∈R d l ),K头注意力机制可以表示为: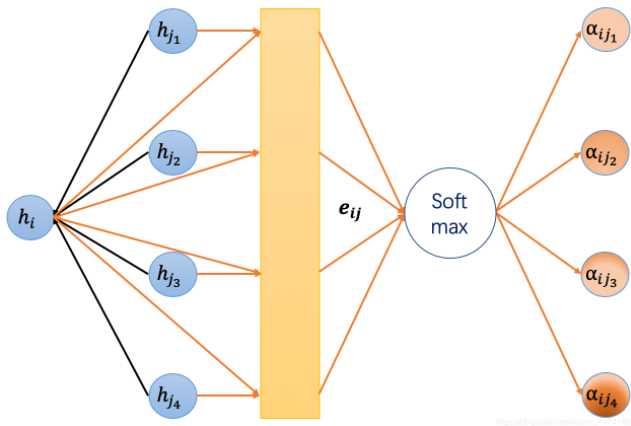
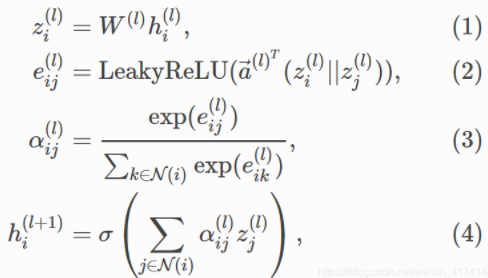

公式参数:*
W:表示一个加权矩阵,用来实现一个可学习的线性变换从h l − 1 h^{l-1}h l −1到h l h^{l}h l
N m N_m N m :节点M邻居节点个数
||:表示连接运算
α m n k α_{mn}^k αm n k :表示第k个注意机制计算的归一化注意系数
e i j ( l ) e_{ij}^{(l)}e i j (l ):节点i和邻居节点j的相似系数
α m n α_{mn}αm n 的定义如下:

GAT采用多头注意机制,提高了自我注意带来的效益。GATs模型的参数如下所示。
- 前两个图注意力层:K=3
- 最后一个图注意力层:K=1
- 全连接层神经元数量:128
一对蛋白质空间图G i G_i G i 和G j G_j G j 最终得到的结构特征向量为F g i ∈ R 1 ∗ 128 F_g^i∈R^{1128}F g i ∈R 1 ∗1 2 8和F g j ∈ R 1 ∗ 128 F_g^j∈R^{1128}F g j ∈R 1 ∗1 2 8。
; 3.4 预测模块
们将序列编码器的输出(即F s i F_s^i F s i 和F s j F_s^j F s j )和结构特征嵌入F g i F_g^i F g i 和F g j F_g^j F g j 进行组合,组合规则如下,其中 λ是一个可学习的参数。

连接F o i F_o^i F o i 和F o j F_o^j F o j ,即[F o i F_o^i F o i ;F o j F_o^j F o j ]并获得输出为:

公式参数:
MLP:为三层堆叠的全连接层
激活函数:作者通过在酵母数据集的实验发现LeakyReLU差于RELU激活函数,故采用RELU作为激活函数。

公式参数:
D:训练数据集中蛋白质-蛋白质样本的总数
σ:表示sigmoid机会函数
- 结果
4.1 种内数据集上的性能比较
4.1.1四个种内数据集性能比较
在种内数据集上得到的 5倍交叉验证结果如下表所示。

结论:
- 准确度: 在e.c oli数据集C和线虫数据集和D. melanogaster数据集均大于99%。
- MCC:均高于95%
; 4.1.2与其他算法比较
基于酵母数据集的方法,并将我们的模型与9个最先进的模型进行了比较,对表3中的所有模型进行训练,直到在5倍交叉验证设置下收敛。
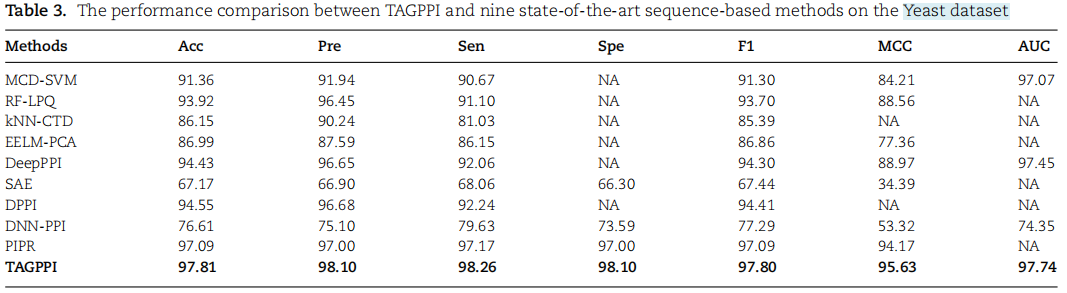
下表中提供了统计显著性检验结果。结果表明TAGPPI明显优于其他方法和TextCNN模块。
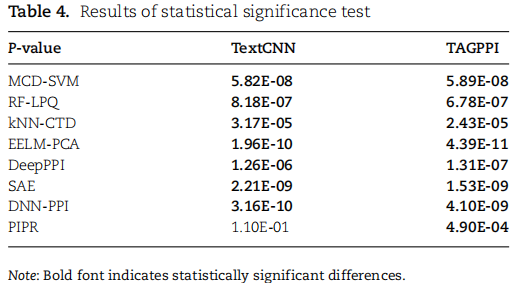
结论:
TAGPPI明显优于其他方法和TextCNN模块
4.2 多物种数据集的性能比较
4.2.1 不同阈值的序列同一性比较
对多物种数据集进行评估的目的是展示TAGPPI在不同序列一致性标准下预测不同物种蛋白质相互作用的能力,下表的结果表明TAGPPI在不同序列一致性阈值下均表现良好。

结论:
模型在测试时的精度和特异性都在99%以上
; 4.2.2 TAGPPI与PIPR方法比较
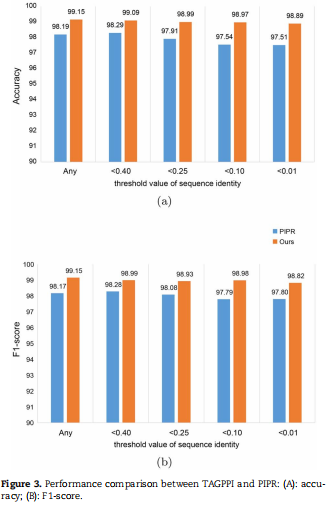
结论:作者的方法TAGPPI的ACC和F1均高于PIPR方法
4.3 多类别数据集性能比较
进一步评估了TAGPPI在PPI类型预测任务中的能力。并与SCNN和PIPR在多类数据集上的性能进行了比较。10倍交差验证的结果如下表所示。

结论:除F1外作者的模型的效果好于SCNN和PIPR。
; 4.4 消融实验
4.4.1TextCNN和图注意层的影响
为了调查我们模型中每个组件的有效性,我们对酵母数据集进行了消融研究。如表7所示,检验TextCNN和图注意层的对实验结果的影响,如下表所示。

结论:TextCNN和图注意层的多重组合比单独使用效果更好
评估了TAGPPI提高的准确性的统计意义。统计学意义的比较(p值
局部特征和结构特征比例的影响:

结论:λ的值为0.3和0.5能取得更好的结果。
; 4.4.2 残基嵌入的影响
利用one-hot编码、物理化学特征和PIPR嵌入方法对三种嵌入特征的嵌入性能进行了评价。将SeqVec与上述三种嵌入特征在酵母数据集上的PPIs预测任务中进行了比较。还将one-hot编码和物理化学特征向量串联融合特征进行比较,如下图所示。
- one-hot编码:利用20种氨基酸进行编码
- 物理化学特征:12种复合氨基酸的理化性质,在12种性质中,疏水性和极性分别按两种不同的尺度计算每种氨基酸的14维物理化学特征向量。
- PIPR的氨基酸嵌入:训练前和物理化学性质的串联。
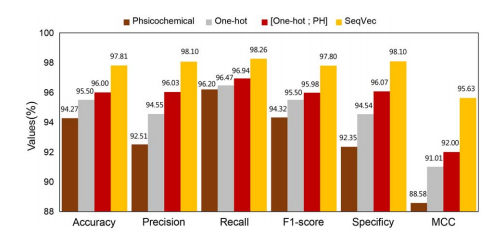
结论: SeqVec嵌入的性能最好。
4.5 超参数的分析
4.5.1批处理大小的影响
如下图所示,随着训练历元的增加,accuracy和precision的值逐渐提高。我们观察到,与批次16、32、64相比,批次8的精度值和精度值的提升趋势不是很明显,在epoch 10前后非常优秀,即使在训练epoch 50之后,批处理大小8也无法实现。
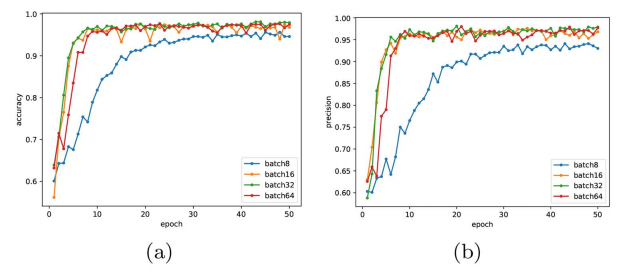
结论:综合考虑时间和精度,batchs_size设置为32
; 4.5.2 分类维度的选择
设计了三层堆叠全连通的二值分类器,并分别实现了Relu激活功能。尺寸表示第一个完全连接层中的神经元数量。第二层完全连接的神经元数量是第一层的一半。分类器的最后一个全连接层只有一个神经元。
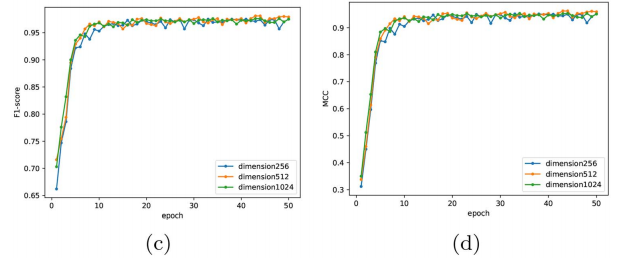
结论:第一个全连接层的输出维度设置为512。
- 结论
提出了一种新的端到端深度学习方法TAGPPI来预测ppi。我们使用卷积结构和GATs同时从氨基酸序列和接触图中提取特征,以描述蛋白质的空间结构。实验评估表明,与其他基于序列的方法相比,我们的模型在PPI预测方面是非常成功的。第一个应用图形的模型学习蛋白质接触映射到PPI任务。
-
创新点
-
提出了一种深度学习框架TAGPPI,将蛋白质的序列特征和预测结构信息整合到蛋白质相互作用(protein-protein interaction, PPI)预测任务中,利用AlphaFold预测数据库构建的联系图,在基于序列和基于结构的方法之间架起了一座桥梁。
- 实验结果表明该方法优于现有的基于序列的方法
-
蛋白质表示学习方法的新策略可以应用于其他任务
-
启发
-
可学习就别手动调参,局部特征和结构特征的比例系数λ的设置。
- AlphaFold预测蛋白质的结构信息。
- 采用多尺度的TextCNN。
- 消融实验验证算法模块作用的同时提升工作量。
Original: https://blog.csdn.net/wangpan007/article/details/122499024
Author: wangpan007
Title: 论文解读:学习蛋白质的空间结构可以提高蛋白质相互作用的预测
原创文章受到原创版权保护。转载请注明出处:https://www.johngo689.com/528046/
转载文章受原作者版权保护。转载请注明原作者出处!